


The advent of molecular pathology has fueled unprecedented advances in the diagnosis and understanding of melanocytic tumors. These advances, however, have also generated concepts that may be difficult to grasp for clinical practitioners, who are not always conversant with the array of genetic techniques employed in the laboratory. These same practitioners, however, are being increasingly called on to provide treatments that are often based on the latest molecular findings for melanocytic tumors. We review the most recent concepts in the pathway classification of melanocytic tumors, including intermediate lesions known as melanocytomas. We examine the genetic and molecular techniques used to study these tumors, look at where they overlap, and discuss their limitations and some of the most difficult-to-interpret results.
Con el desarrollo de la enfermedad molecular, el diagnóstico y la comprensión de los tumores melanocíticos ha experimentado un avance descomunal en los últimos años. Esto ha significado la aparición de conceptos de difícil asimilación en el mundo clínico, el cual no siempre está en contacto directo con las técnicas genéticas de laboratorio. Al mismo tiempo, sin embargo, al clínico se le está exigiendo una terapéutica basada en muchas ocasiones en los hallazgos moleculares más recientes de un tumor melanocítico. El presente artículo explora los conceptos moleculares más recientes de la clasificación en rutas patogénicas de los tumores melanocíticos, incluidas las formas intermedias conocidas como melanocitomas, y repasa las técnicas auxiliares usadas en el estudio de estos tumores, discutiendo los resultados más complejos, sus limitaciones y sus solapamientos.
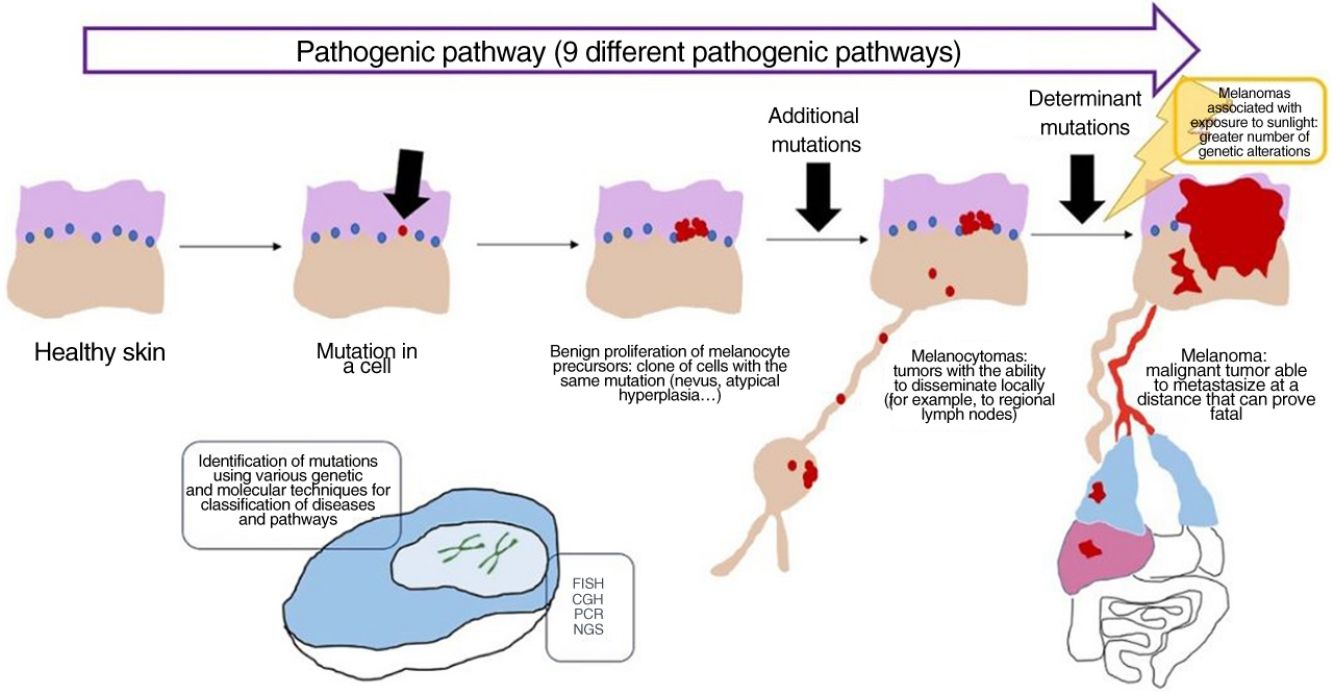
In recent decades, genetic and molecular techniques have considerably increased our knowledge of the etiology and pathogenesis of melanoma, thus enabling us to decipher a complex code in which the duad comprising benign nevus and malignant melanoma as the only 2 options has given way to a spectrum involving several intermediate stages.
These techniques have also shown us that there is not only 1 type of melanoma, but rather various types that can develop via different and independent pathogenic routes.
More precise identification of the types lying between nevus and melanoma has been accompanied by conceptually difficult diagnoses of conditions that cannot be considered benign or malignant in absolute terms but must be seen as an intermediate progression to melanoma. A better understanding of the latter was achieved with the incorporation of sentinel node biopsy and evidence for melanocytic tumors that disseminate locally (e.g., to the lymph nodes) without proving fatal.
Description of such complex concepts has necessitated the resurrection of terms considered almost extinct in the literature, such as melanocytoma. Hence the need for dermatologists to understand the histopathologic status of such an ongoing and dynamic landscape.
Our objective was to explain and update some of these concepts as seen through the eyes of the pathologist and adapt them to the practical clinical needs of the dermatologist.
Melanocytic Tumors Are the Result of Genetic AlterationsAs with so many other tumors, melanocytic tumors, whether benign or malignant, result from various types of genetic alterations.
Nevi are usually caused by a single mutation in a melanocyte, which in turn leads to benign clonal expansion. Acquired nevi result from mutations in melanocytes that have generally reached the epidermis, leading initially to junctional nevi, which become compound and then intradermal. Most acquired nevi are characterized by a mutation at codon 600 in the BRAF gene.1,2
In the case of congenital nevus, on the other hand, the mutation arises at some time during the migration of the melanocyte to the epidermis from the neural crest. The closer the melanocyte is to the epidermis, the smaller the nevus, whereas if the mutation arises early during migration, the congenital nevus can become quite extensive, affecting various areas of the body. In contrast with acquired nevi, congenital nevi are usually characterized by mutations in the NRAS gene, very often at codon 61.3,4
Exposure to sunlight is one of the most important factors – although not the only one – in the pathogenesis of melanoma,5–7 with more genetic aberrations being observed in tumors that develop as a result of intense, prolonged exposure to sunlight.8–13 On the other hand, melanomas that may have little or no association with sunlight (e.g., acral melanoma) have a low mutational burden.13–15 UV-A radiation is less carcinogenic than UV-B radiation: the former induces oxidation of guanine in DNA, whereas the latter generates oncogenic cyclobutane dimers.16,17
Decades of examining melanomas through an optical microscope have revealed the broad morphological variety of these tumors, which can present as various lesions, for example, superficial spreading melanoma, nodular melanoma, spitzoid melanoma, and acral melanoma. However, despite these variations, the genomic pathogenesis of melanoma is limited, affecting a small group of molecular pathways. The main pathways are as follows18–23:
- 1.
The MAPK/ERK pathway (MAP kinases [MAPK], also known as ERK), which comprises several kinases regulated by extracellular factors. MAPK includes MAP2K1, MAP3K, MAP2K, and TRK. Along this pathway, the signal is transmitted to the nucleus via proteins such as RAS and RAF and transcription factors such as MYC.
- 2.
The phosphatidylinositol-3-kinase PI3K/AKT/mTOR pathway.
- 3.
The oncogenic p53 pathway.
- 4.
The MITF pathway.
- 5.
The NFkB pathway.
- 6.
The cell cycle pathway (G1/S transition).
- 7.
The WNT pathway (regulation of apoptosis).
- 8.
The chromatin remodeling pathway (SWI/SNF).
In genetic terms, some melanocytic tumors can be distinguished from others by the mechanisms through which these pathways are altered. For example, the MAPK pathway can be altered by mutations in BRAF, NRAS, and NF124–26 (frequent in common nevus and melanoma associated with low-dose sun exposure), although it can also be altered by mutations in HRAS or by kinase gene fusion in the case of Spitz nevus.27
Concept: Pathogenic PathwaysWhile mutations can be found in nevi, they are often insufficient to render a cell invasive or metastatic. With melanocytic nevi, on the other hand, growth is limited owing to senescence, which causes a cell to fail in its attempt to re-enter the cell cycle, thereby preventing mitosis.28 Senescence is induced by various mechanisms, thus hampering the development of uncontrollable tumors. The tumor-suppressor genes p53 and p16 are among the most well-known mechanisms.29
If a nevus undergoes additional genetic and epigenetic changes, it can escape the mechanisms of senescence and become an intermediate lesion, with a low or high risk of progression, or a melanoma. This interpretation of the nature of melanocytic tumors has resulted in the identification of several pathogenic pathways.30–32 Each pathway has a benign nevic counterpart or benign hyperplastic precursor, as well as intermediate types of malignancy, until the development of melanoma. Some pathways are associated with exposure to sunlight as the cause, whereas others are not.33 This model remains largely hypothetical, since the benign precursors of some entities have yet to be categorized.
The pathways are not always activated during the initial phase. For example, melanocytoma can develop de novo, despite being considered an intermediate stage between nevus and melanoma. In fact, for many years the general axiom was that most melanomas develop de novo and that only a minority originated from a nevus.34,35 This axiom requires some clarification, since the most common melanoma in our setting, lentigo maligna melanoma, arises from a series of previous mutations in situ (even if they are not “nevic”), such as lentigo maligna or similar entities. De novo onset of a melanoma with direct infiltrating properties is difficult to account for based on genetics, since it would require a build-up of sudden, catastrophically occurring genetic events, such as chromothripsis or chromoplexy,36 both of which are uncommon. Some authors provide an alternative explanation, suggesting that histologically normal melanocytes could, on the other hand, have initial mutations that do not induce proliferation (e.g., in CDKN2A or TERT promoter mutations).36,37 Moreover, other authors indicate that our knowledge of this process could be biased: the absence of nevus remnants when a melanoma is removed does not necessarily mean that the melanoma did not arise from a nevus, since the growth of the melanoma or the immune response to this could have destroyed the initial nevus remnants.36
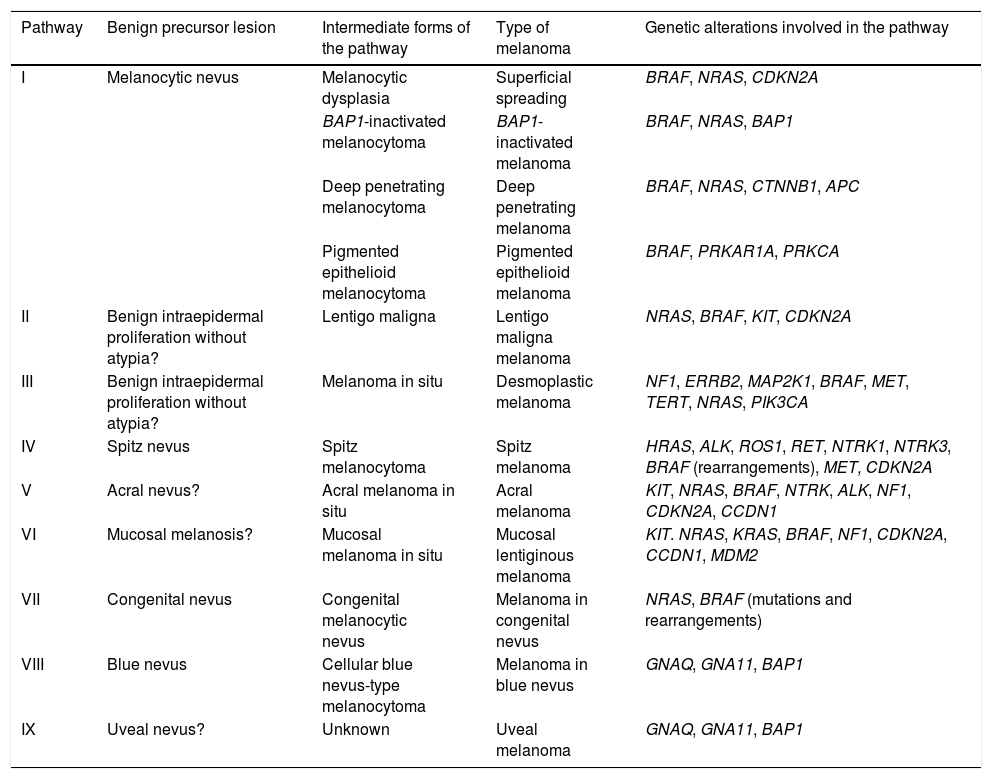
The pathways recognized by the World Health Organization (WHO) are set out below33,38 (Table 1).
Development of the Different Types of Melanoma: Simplified Pathogenic Pathways Identified by the World Health Organization.
| Pathway | Benign precursor lesion | Intermediate forms of the pathway | Type of melanoma | Genetic alterations involved in the pathway |
|---|---|---|---|---|
| I | Melanocytic nevus | Melanocytic dysplasia | Superficial spreading | BRAF, NRAS, CDKN2A |
| BAP1-inactivated melanocytoma | BAP1-inactivated melanoma | BRAF, NRAS, BAP1 | ||
| Deep penetrating melanocytoma | Deep penetrating melanoma | BRAF, NRAS, CTNNB1, APC | ||
| Pigmented epithelioid melanocytoma | Pigmented epithelioid melanoma | BRAF, PRKAR1A, PRKCA | ||
| II | Benign intraepidermal proliferation without atypia? | Lentigo maligna | Lentigo maligna melanoma | NRAS, BRAF, KIT, CDKN2A |
| III | Benign intraepidermal proliferation without atypia? | Melanoma in situ | Desmoplastic melanoma | NF1, ERRB2, MAP2K1, BRAF, MET, TERT, NRAS, PIK3CA |
| IV | Spitz nevus | Spitz melanocytoma | Spitz melanoma | HRAS, ALK, ROS1, RET, NTRK1, NTRK3, BRAF (rearrangements), MET, CDKN2A |
| V | Acral nevus? | Acral melanoma in situ | Acral melanoma | KIT, NRAS, BRAF, NTRK, ALK, NF1, CDKN2A, CCDN1 |
| VI | Mucosal melanosis? | Mucosal melanoma in situ | Mucosal lentiginous melanoma | KIT. NRAS, KRAS, BRAF, NF1, CDKN2A, CCDN1, MDM2 |
| VII | Congenital nevus | Congenital melanocytic nevus | Melanoma in congenital nevus | NRAS, BRAF (mutations and rearrangements) |
| VIII | Blue nevus | Cellular blue nevus-type melanocytoma | Melanoma in blue nevus | GNAQ, GNA11, BAP1 |
| IX | Uveal nevus? | Unknown | Uveal melanoma | GNAQ, GNA11, BAP1 |
Pathway I (low cumulative solar damage [CSD]): Superficial spreading melanoma, in which 90% of melanocytic tumors harbor mutations in the BRAF gene, with the most frequent being V600E. Mutations in NRAS are much less frequent.
Various routes lead to this type of melanoma:
- -
Low-grade dysplasia, followed by high-grade dysplasia.
- -
Inactivation of the BAP1 gene, followed by additional mutations.
- -
Deep penetrating nevus, followed by additional mutations.
- -
Epithelioid melanocytoma, followed by additional mutations.
In this pathway, it is not uncommon during the phases close to malignancy for there to be loss of p16 due to alterations in the CDKN2A gene.39 However, evaluation of p16 involves a series of considerations. While p16 is considered a marker of senescence and, therefore, reassuring when expressed,40–42 it is not, however, expressed in normal melanocytes. It may be expressed in response to cellular stress.43 Some common nevi may express little p16 without this indicating inactivation of CDKN2A.
Pathway II (high-CSD): This pathway culminates in lentigo maligna melanoma, which first develops in lentigo maligna or in other types of melanoma in situ.
Pathway III (also high-CSD): Desmoplastic melanoma, which first appears as melanoma in situ.
Pathway IV: Spitz melanoma, which first appears as benign Spitz nevus.44,45 It usually involves HRAS-activating mutations (with[out] concomitant amplifications of 11p, which is where HRAS is located), rearrangements of receptor tyrosine kinase genes (ROS1, ALK, MET, NTRK1, NTRK3, and RET), or rearrangements of the serine-threonine kinase genes involved in the MAPK pathway (BRAF, RAFF1, MAP3K8).37,46 Kinase rearrangements are mutually exclusive (the presence of one excludes the others). BRAF rearrangements do not involve mutations in the gene (which, in contrast, are common in pathway I). Numeric changes (e.g., amplifications) may sometimes occur in the genes adjacent to the rearranged areas of Spitz tumors, even benign tumors, without these necessarily carrying a greater risk of progression.
One curious characteristic of the Spitz tumor pathway is inactivation of CDKN2A (often the only genetic alteration), with loss of p16 expression, which would be of concern in melanocytic tumors in any other pathway.47 Therefore, malignant transformation requires other mutations in addition to inactivation of CDKN2A, for example, TERT promoter mutations.
Another common characteristic in this group is that malignant lesions can conserve previous mutations from benign precursor lesions. Therefore, while HRAS mutations are typical of Spitz nevi, demonstrating their presence is no guarantee of a benign lesion, since they can also be found in Spitz melanoma.48,49 However, given that HRAS mutations are found in less than 1% of melanomas,37 their presence in a Spitz tumor is reassuring.
Various attempts have been made to correlate the genetic alterations observed in Spitz tumors with histopathology findings in hematoxylin–eosin staining,50 starting with the suggestion that HRAS mutations activate the PI3K/AKT/mTOR pathway more vigorously than BRAF or NRAS mutations and, therefore, induce larger, less pigmented tumor cells than those seen in common or congenital nevi.37 While these correlations are not always easy to evaluate, we know, for example, that HRAS mutations are associated with the following: desmoplasia; rearrangements in MAP3K8, with a predominance of epithelioid cells, multinucleate cells, and ulceration; rearrangements in RET, with tumor shadow on imaging and poorly cohesive cell nests; rearrangements in NTRK3, with pigmented tumors; rearrangements in NTRK, with the presence of pseudorosettes; rearrangements in ROS1, with abundant Kamino bodies; and rearrangements in ALK, with large dome-shaped or polypoid tumors, which are often fasciculate in, sometimes with a prominent myxoid deposit. However, these correlations are not complete. Thus, for example, a desmoplastic phenotype may not be associated with an HRAS mutation or 11p amplification and, in contrast, be associated with kinase fusions, as is the case with ROS1.
Rearrangements in BRAF, unlike fusions in other kinases, seem to be terminal events in the pathway, that is, they are more associated with the malignant part of the spectrum, since they have been seen mainly in Spitz melanomas, in Spitz melanocytomas, and, albeit to a much lesser extent, in Spitz nevus.
Pathway V: Acral melanoma, which may begin as acral nevus or in situ malignant precursor acral lesions.
Pathway VI: This pathway starts in dysplastic melanosis and leads to mucosal lentiginous melanoma. We do not know if nondysplastic melanosis could be the origin of the pathway.
Pathway VII: Melanomas arising on congenital nevi. Growth nodules may appear on congenital nevus owing to mutations occurring in addition to nevus mutations. Therefore, they have a low-to-intermediate risk of progressing to melanoma. The pathway leads to melanoma on congenital nevus. Melanocytic lesions on pathway VII contain NRAS hotspot mutations and, less frequently, BRAF mutations.
Pathway VIII: Melanomas arising in blue nevi, passing through cellular blue nevus (included in low- to intermediate-risk melanocytomas that progress to melanoma), atypical cellular nevus, and, eventually, melanoma on blue nevus, which is an uncommon entity. Pathway VIII arises mainly owing to oncogenes that activate the G-α-q pathway, with the most common genetic alteration being activating mutations in GNAQ or in GNA11. The last stages of the journey to malignancy may be characterized by mutations of the BAP1 tumor-suppressor gene.
Pathway IX: Uveal melanoma. The histologic stages preceding transformation to melanoma are unknown. The pathway may present mutations in the tumor suppressor gene BAP1.
This outline of the pathogenic pathways makes it easy to explain why nevi are more numerous in clear-skinned persons and in persons with melanoma susceptibility syndromes, since their melanocytes are more predisposed to mutation. It also enables us to see why episodes of intermittent exposure to sunlight during infancy are associated with a greater number of nevi and why a larger number of nevi implies a greater risk of melanoma.
It is important to note that, according to the WHO, not all melanocytic tumors carry the same risk of progression to melanoma. Some are classed as low- to intermediate-risk (e.g., atypical melanosis, melanocytoma in congenital nevus, deep penetrating nevus, and blue nevus-type melanocytoma), whereas others are considered intermediate- to high-risk (e.g., BAP1-inactivated melanocytoma, deep penetrating melanocytoma, pigmented epithelioid melanocytoma, lentigo maligna, melanoma in situ [whether acral, mucosal, or congenital], and atypical cellular blue nevus). This also explains why some lesions that appear less spectacular from a histopathologic standpoint (e.g., acral melanoma in situ, sometimes with very subtle histopathologic changes that are difficult to differentiate from nevus) progress rapidly to invasive melanoma if they are not correctly diagnosed on time.
Concept: MelanocytomaThe term melanocytoma has been consolidated in the WHO classification as referring to melanocytic tumors that occupy an intermediate place in the progress of a lesion along a pathogenic pathway from benign to malignant.51 Given that this type of tumor has multiple driver mutations, it has been classified separately from nevus. Melanocytoma may occasionally present alongside common melanocytic nevus, indicating that a new melanocytoma clonal population has arisen within a nevus because of additional rearrangements or mutations. However, since melanocytomas sometimes occur de novo, the previous step of common nevus is not always necessary.
Many melanocytomas can disseminate to regional lymph nodes without further metastasis. The WHO divides them into 2 groups at risk of progression to melanoma, namely, low- to intermediate-risk and intermediate- to high-risk.
At present, the most common types of melanocytoma are as follows:
- -
BAP1-inactivatedmelanocytoma52: This melanocytic lesion falls within the pathogenic pathway of intermittent CSD melanoma (pathway I). Tumors in this group are characterized by biallelic inactivation of the tumor suppressor gene BAP1, mostly through association with a truncating mutation in one of the alleles of chromosome 3 (locus 3p21) and complete or partial loss of the other. This represents an intermediate step between Wiesner nevus (also known as BAPoma) and melanoma carrying inactivating BAP1 mutations. The concept of BAP1-inactivated melanoma is essentially based on genetics, and histology findings alone do not always make it possible to distinguish clearly between BAPoma and melanocytoma. Therefore, some authors have suggested grading atypia to provide additional information in this type of diagnosis.52 Similarly, BAP1-inactivated melanoma usually presents a sufficient number of histopathologic alterations to suggest this diagnosis. However, such progression can be ruled out by the absence of genetic alterations typical of melanoma in the case of a BAP1-inactivated tumor and a specific grade of atypia. Inactivation of BAP1 can coexist with mutations in NRAS and BRAF. Therefore, and in contrast with what was previously thought, this is not a variant of a Spitz tumor. In most cases, gene inactivation can be demonstrated using immunohistochemistry, which reveals a loss of nuclear staining.53 However, immunohistochemistry does not identify cases with nonsense mutations, where nonfunctional protein continues to be produced.
- -
Some individuals have a germline BAP1 mutation in one of the alleles, which manifests as multiorgan tumor predisposition syndrome,54 with a greater risk of mesothelioma, renal carcinoma, cholangiocarcinoma, basal cell carcinoma, and, frequently, cutaneous and uveal melanoma. The WHO considers the risk of BAP1-inactivated melanocytoma transforming to melanoma to be medium-to-high, although this is not consistent with experience reported elsewhere.37BAP1-inactivated melanocytoma does not generally involve mitosis or only occasionally shows mitosis. A high mitotic index is a strong indicator that malignant transformation has already occurred. It is important to remember that BAP1 mutations are not exclusive to this type of melanocytoma and may also be seen in other types of melanocytic tumor; therefore, immunohistochemistry that is negative for BAP1 does not guarantee a diagnosis of this type of melanoma. Moreover, since BAP1 mutations are characteristic of late phases (of malignant transformation) in other pathogenic pathways (e.g., uveal melanoma55 or blue nevus–type melanoma56), they should be addressed with caution and give cause for concern in these settings.
- -
Deep penetratingmelanocytoma57: Deep penetrating melanocytoma also belongs to pathway I and is an intermediate lesion, between deep penetrating nevus and melanoma in a deep penetrating nevus. Histologically, melanocytoma, unlike nevus, has atypical characteristics, such as large size, asymmetry, melanocytes arranged in sheets, mitosis, and severe cytologic atypia. However, melanocytoma does not yet show the genetic changes that are considered typical of melanoma; therefore, in the case of more atypical melanocytoma, the absence of progression to melanoma should be demonstrated using special techniques (see below). This group harbors mutations (generally point mutations) that activate β catenin, a process that can be demonstrated by positive immunohistochemistry findings for the β catenin marker58 or, alternatively, for LEF1 (the nuclear receptor of β catenin).59 In the case of common nevus, positive findings for β catenin are only observed in melanocytes close to the epidermis and adnexa, whereas in deep penetrating melanocytoma, β catenin stains the cytoplasm of the tumor melanocytes uniformly and diffusely. It may also intensely stain the nucleus. β Catenin is not the only mechanism by which deep penetrating melanocytoma is generated. It can also arise, for example, from the biallelic loss of function in the adenomatous polyposis coli (APC) gene.57 The WHO considers the risk of transformation of deep penetrating melanocytoma to melanoma to be medium-to-high, as demonstrated in cases where the tumor proved fatal after progression.60
- -
Pigmented epithelioid melanocytoma: Pigmented epithelioid melanocytoma also belongs to pathway I and usually carries BRAF mutations. It is an indolent melanocytoma that, despite invading a large number of lymph nodes, is not usually characterized by distant metastasis. No fatal cases of pigmented epithelioid melanocytoma have been reported. It is not genetically related to blue nevus, despite sharing similar histologic findings.
- -
This group contains a variant known as PRKAR1A-inactivated melanocytoma61,62 (wrongly referred to in the past as epithelioid blue nevus), which has been associated with Carney complex.63 Inactivation of PRKAR1A can be demonstrated by negative immunohistochemistry findings for anti-PRKAR1A antibody. In parallel, other pigmented epithelioid melanocytomas are characterized by combinations of BRAF mutations with inactivation of PRKAR1A. Lastly, other tumors in this group present genetic alterations such as mutations in GNAQ and MAP2K1 and even fusions in PRKCA. The question of whether these tumors can continue to be included in this group of indolent melanocytomas or whether they constitute another type of melanocytic tumor belonging to another pathway has yet to be answered. The WHO considers that the risk of transformation of pigmented epithelioid melanocytoma to melanoma is medium-to-high.
- -
Spitzmelanocytoma45: Spitz melanocytoma belongs to pathway IV, which ranges from Spitz nevus to Spitz melanoma. Spitz melanocytoma presents alterations of the CDKN2A gene, which codes for p16 and p14.44 Therefore, whereas the loss of expression of p16 is a cause for concern in many melanocytic tumors, especially those belonging to pathway I, it is common in the Spitz pathway. Inactivation of CDKN2A frequently results from homozygous deletion, although it can also arise from a truncating mutation in an allele, followed by a loss of heterozygosity, often due to loss of part of chromosome 9. Given that some tumors may retain a copy of CDKN2A that remains active, fluorescence in situ hybridization does not provide information in such cases, and the study must be complemented with immunohistochemistry for detection of p16 (which remains positive). p16 may be cytoplasmic or nuclear and is often seen as a mosaic pattern, that is, alternative positive and negative cells. Spitz melanocytoma frequently invades lymph nodes, although it is generally benign, with a low recurrence rate. The WHO considers that its risk of transformation to melanoma is low-to-medium. Whereas Spitz melanocytoma shows a heterozygous deletion of CDKN2A, a homozygous deletion should lead to a high suspicion of Spitz melanoma. Additional mutations, such as TERT promoter mutations, point to the possible progression of Spitz melanocytoma to malignant disease.
- -
Melanocytoma in congenitalnevus64: Melanocytoma in congenital nevus is an intermediate nodular lesion that appears on a congenital nevus and thus belongs to pathway VII. According to the WHO classification, this melanocytoma is synonymous with the term nodule in congenital nevus. Despite its alarming histologic appearance, melanocytoma in congenital nevus is usually characterized by losses or gains in whole chromosomes, in contrast with melanoma, which is characterized by numerical alterations of chromosomal segments.65
- -
The WHO considers that the risk of transformation to melanoma is low-to-medium.
- -
Blue melanocytoma: In the past, blue melanocytoma was also known as cellular blue nevus.66 As a tumor from pathway VIII, it usually harbors mutations that activate the G-α-q pathway (mutations in GNAQ). In contrast with deep penetrating melanocytoma, where β catenin is expressed in the nucleus, blue melanocytoma expresses this marker in the membrane. The WHO considers that the risk of transformation to melanoma is low-to-intermediate.
The WHO recommends complete excision with wide margins (5mm) for melanocytic tumors with a high risk of progressing to melanoma; however, not all authors consider the risk of progression of the various types of melanocytoma to be a proven fact.51
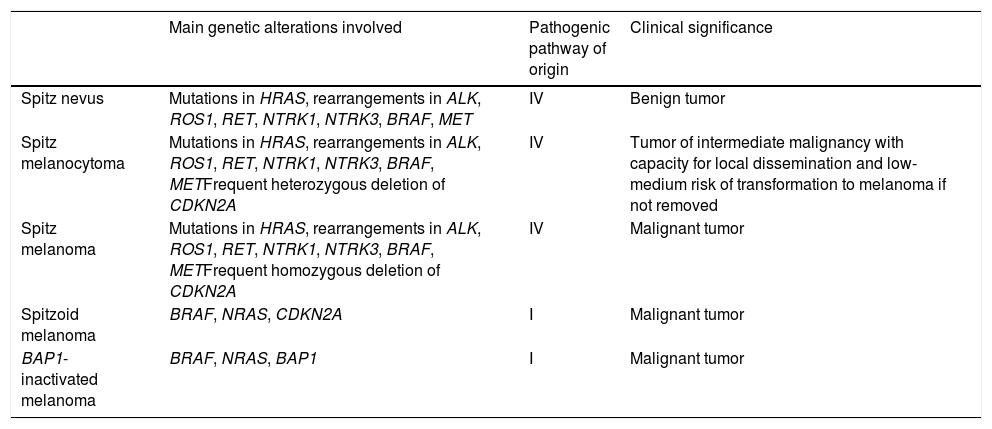
Spitzoid Melanoma Is Not the Same as Spitz Melanoma. Spitz Tumor Is Not the Same as Spitzoid Tumor (Table 2)Spitz melanoma is the end of pathway IV, which starts as Spitz nevus and passes through Spitz melanocytoma (Table 2).
Differences Between the Main Melanocytic Tumors With a Spitzoid Phenotype.
| Main genetic alterations involved | Pathogenic pathway of origin | Clinical significance | |
|---|---|---|---|
| Spitz nevus | Mutations in HRAS, rearrangements in ALK, ROS1, RET, NTRK1, NTRK3, BRAF, MET | IV | Benign tumor |
| Spitz melanocytoma | Mutations in HRAS, rearrangements in ALK, ROS1, RET, NTRK1, NTRK3, BRAF, METFrequent heterozygous deletion of CDKN2A | IV | Tumor of intermediate malignancy with capacity for local dissemination and low-medium risk of transformation to melanoma if not removed |
| Spitz melanoma | Mutations in HRAS, rearrangements in ALK, ROS1, RET, NTRK1, NTRK3, BRAF, METFrequent homozygous deletion of CDKN2A | IV | Malignant tumor |
| Spitzoid melanoma | BRAF, NRAS, CDKN2A | I | Malignant tumor |
| BAP1-inactivated melanoma | BRAF, NRAS, BAP1 | I | Malignant tumor |
This route involves typical genetic alterations of the Spitz family, such as the absence of mutations in BRAF, NRAS, and NF1 and the frequent presence of rearrangements in kinase genes or HRAS mutations. Since kinases are not usually expressed in healthy mature melanocytes and kinase fusion usually involves their expression, these fusions can be detected using immunohistochemistry based on antikinase antibodies. However, it is important to remember that not all fusions are equally conducive to expression of the corresponding kinase. As for ALK and ROS1 in particular, positive findings in immunohistochemistry can vary, ranging from diffuse cytoplasmic to dot-like. In fact, staining with ROS1 is usually weak. In the case of NTRK (1 and 3), expression is usually so intense that moderate, weak, or patchy expression should be considered doubtful because some isoforms of NTRK are expressed in healthy melanocytes and NTRK can be expressed in non-Spitz melanocytic tumors (e.g., blue nevus and deep penetrating nevus) owing to overexpression of ALK via other molecular mechanisms. It is also important to take into account the nuclear expression of NTRK, which is more common in Spitz tumors with NTRK3 fusions than in those with NTRK1 fusions. When applying immunohistochemistry to assess kinase expression, staining of macrophages should not be misinterpreted as positive. In the case of other kinases (BRAF, RAF1, and MAP3K8), immunohistochemistry seems to be of little use for predicting fusions.36
Staining with hematoxylin–eosin shows some melanocytic tumors (both benign and malignant) to have spitzoid features, although they are not Spitz tumors, because they do not follow the Spitz pathogenic pathway and therefore do not show the genetic alterations that are characteristic of these tumors. An example can be seen in adult spitzoid tumors that follow pathway I and frequently harbor mutations in BRAF, NF1, or NRAS. In fact, given that malignant transformation of a benign Spitz tumor to melanoma is rare, Spitz melanoma is not a frequent finding. Melanomas with a spitzoid appearance are much more common.
One group of spitzoid melanomas that requires special mention is BAP1-inactivated tumors. Until recently, these melanocytic lesions were erroneously included in the Spitz tumor spectrum. It was no less surprising, therefore, that most had mutations in BRAF or RAF1,67 a perfectly understandable finding today, since we know that these melanomas belong to pathway I (low-CSD) and that mutations in BAP1 are yet another event in transformation to melanoma. Consequently, in assessment with hematoxylin–eosin, most BAPomas correspond to combined nevi, with a mutation in BAP1 in a focal nodule of the melanocytoma, in the setting of a common nevus with mutations in BRAF. The melanocytoma nodule shares the same BRAF mutation as other nevi, because the mutation in BAP1 arose after that in BRAF. These tumors sometimes have the appearance of a spitzoid tumor, albeit with a prominent inflammatory infiltrate. Therefore, many were previously classified as Spitz nevus with halo phenomenon. However, not all spitzoid melanocytic tumors with a prominent infiltrate are BAPomas. Thus, real Spitz tumors with fusions in NTRK1 may be accompanied by a large number of lymphocytes.
Noninterchangeable Terms: Atypical Spitz Tumor Is Not Spitz MelanocytomaFor the same reason, Spitz melanocytoma belongs to the same pathogenic pathway as the Spitz family. By consensus, Spitz melanocytoma must show deletion of CDKN2A with negative findings for p16, albeit with no additional numeric chromosomal aberrations or additional mutations, especially in the TERT promoter region. While the terms Spitz melanocytoma and atypical Spitz tumor have often been used interchangeably in the literature, the term Spitz melanocytoma should be reserved for lesions with the above-mentioned genetic changes.
Furthermore, given their changes in BRAF, NRAS, and BAP1, many nevi that were previously classified as atypical Spitz tumor are now considered other types of melanocytic tumor included in other pathogenic pathways, such as pathway I (low-CSD).
Concept: Special Site NevusStaining with hematoxylin–eosin reveals atypical or worrying characteristics in some melanocytic nevi. However, genetic and molecular studies of these nevi have not identified risk in terms of prognosis or progression to malignancy, thus leading us to believe that histologic changes are the result of the location of these nevi at special sites (e.g., areas of friction or exposure to other agents).68
The main special sites are the breasts, skinfolds, scalp, genitals, and acral areas.68
The histologic changes typical of special site nevi are varied, although they often give cause for concern and include confluence of nests, a certain degree of pagetoid spread, areas of dermal fibrosis, inflammatory infiltrates, loss of melanocytic cohesion in nests, irregular nesting patterns, and melanocytes with hyperchromatic nuclei. Knowledge of potential changes associated with a specific anatomic location is mandatory if we are to avoid overdiagnosis of melanoma in these cases.
Another important consideration with respect to special site nevi is that, although they are characterized by atypia, they are not “dysplastic nevi”, which are understood to be nevi that indicate risk of melanoma in some families,69 and should therefore not be diagnosed as such.
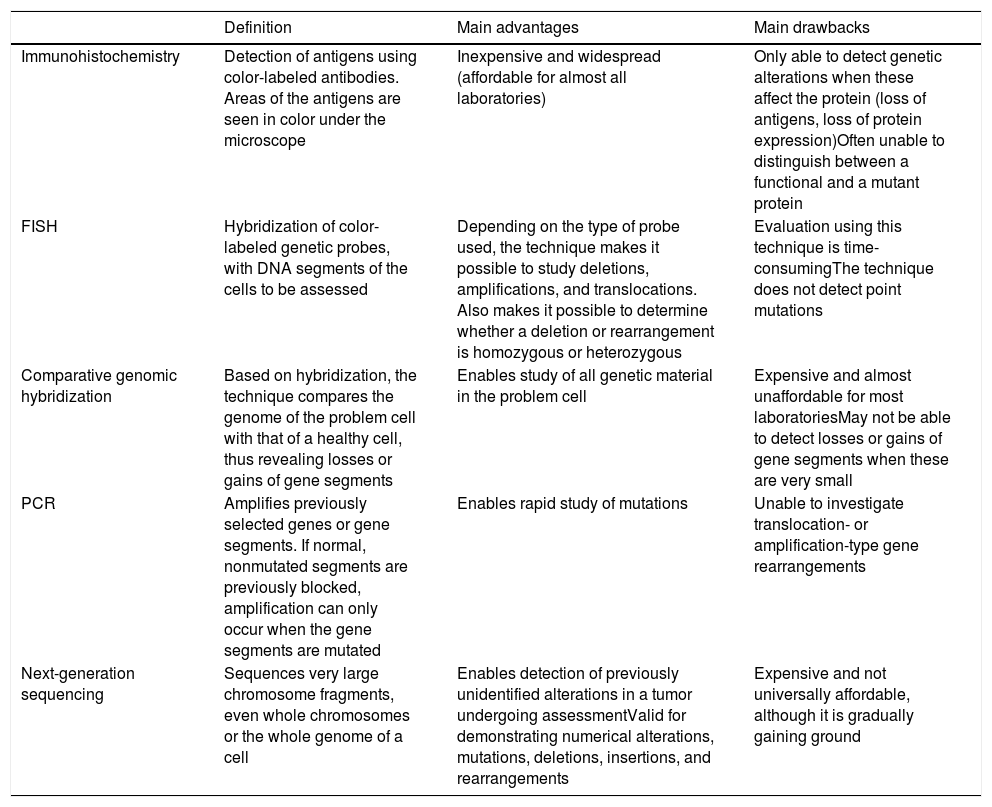
Detection of Genetic Alterations in Clinical Practice: Main Techniques (Table 3)Melanocytic tumors are characterized by genetic alterations from onset, that is, even when they are benign. There may even be epigenetic alterations that inactivate the gene without the need for structural alterations. The latter, therefore, cannot be detected using standard genomic techniques. Genetic alterations are varied, ranging from point mutations to losses of more or less large chromosomal segments, whole arms, whole chromosomes, and duplications of genetic material (parts of chromosomes or whole chromosomes) (Table 3).
Main Additional Techniques Used for Assessment of Melanocytic Tumors.
| Definition | Main advantages | Main drawbacks | |
|---|---|---|---|
| Immunohistochemistry | Detection of antigens using color-labeled antibodies. Areas of the antigens are seen in color under the microscope | Inexpensive and widespread (affordable for almost all laboratories) | Only able to detect genetic alterations when these affect the protein (loss of antigens, loss of protein expression)Often unable to distinguish between a functional and a mutant protein |
| FISH | Hybridization of color-labeled genetic probes, with DNA segments of the cells to be assessed | Depending on the type of probe used, the technique makes it possible to study deletions, amplifications, and translocations. Also makes it possible to determine whether a deletion or rearrangement is homozygous or heterozygous | Evaluation using this technique is time-consumingThe technique does not detect point mutations |
| Comparative genomic hybridization | Based on hybridization, the technique compares the genome of the problem cell with that of a healthy cell, thus revealing losses or gains of gene segments | Enables study of all genetic material in the problem cell | Expensive and almost unaffordable for most laboratoriesMay not be able to detect losses or gains of gene segments when these are very small |
| PCR | Amplifies previously selected genes or gene segments. If normal, nonmutated segments are previously blocked, amplification can only occur when the gene segments are mutated | Enables rapid study of mutations | Unable to investigate translocation- or amplification-type gene rearrangements |
| Next-generation sequencing | Sequences very large chromosome fragments, even whole chromosomes or the whole genome of a cell | Enables detection of previously unidentified alterations in a tumor undergoing assessmentValid for demonstrating numerical alterations, mutations, deletions, insertions, and rearrangements | Expensive and not universally affordable, although it is gradually gaining ground |
Abbreviations: FISH, fluorescence in situ hybridization; PCR, polymerase chain reaction.
FISH enables hybridization of fluorescent probes and chromosomal segments in the melanocytic tumor. Thus, we can detect absence, change of position (e.g., translocations, fusions), and amplifications of these segments. Three special probes are worthy of mention in the diagnosis of difficult-to-classify melanocytic tumors,70,71 namely, probes for the regions CCND1 (11q13), RREB1 (6p25), and MYB (6q23). The combination of all 3 probes has proven successful for differentiating between melanoma and nevus. The results should always be interpreted in the setting of histopathology findings. Thus, for example, a single translocation in the probes mentioned does not automatically indicate a melanoma. Similarly, a negative result for several probes does not guarantee that the tumor is benign.
Comparative genomic hybridization compares the genome of the melanocytic tumor with that of a healthy cell adjacent to the lesion.72 Thus, losses or gains in more or less large chromosomal segments, or even whole chromosomes, can be detected. It is only when the segments are too small that they remain “invisible” in comparative genomic hybridization. While nevi do not usually have alterations in copies of a chromosomal segment (with a few exceptions, such as amplifications of 11p in some Spitz nevi), melanomas are usually characterized by many numerical abnormalities, which include chromosomal segments or whole chromosomes.
Polymerase chain reaction is a technique for amplification of short genome segments that makes it possible to detect mutations in a specific gene. It is based on the following principle: a synthetic polymer similar to DNA is used to block normal segments of the gene under study in such a way that only the mutated segments are amplified in the reaction cycles. The technique has proven very successful for detection of mutations in BRAF and NRAS.73
Protein production is the optimal end result of transcription and translation of a gene. Proteins can be detected using immunohistochemistry, which is based on antibodies against protein antigens. Thus, for example, we can detect cytoplasmic kinases in many Spitz tumors.36 Value can also be attributed to the loss of protein expression, which is a negative finding in immunohistochemistry. The technique is limited by cases of anomalous nonfunctional protein production, where immunohistochemistry continues to be positive. Within the technique of immunohistochemistry, special mention must be made of preferentially expressed antigen in melanoma antibody (PRAME), whose expression in the nucleus should give cause for concern in the case of a histologically atypical melanocytic lesion.74,75 Staining with PRAME should not be evaluated in absolute terms (positive/negative), but rather in relative terms (intensity and percentage of staining). Moreover, it should be interpreted in the context of the histology findings. Applied in this way, it has played a considerable role in the identification of many melanocytic lesions in daily clinical practice. One of its main drawbacks, however, is that of melanocytic tumors with a spitzoid morphology, where the results of PRAME testing are not as good as in other types of melanocytic tumor. In this sense, a recently published article presented the combination of immunohistochemistry for p16 and BRAF V600 as being superior to PRAME testing in spitzoid tumors.76
Techniques for sequencing of short gene segments have been complemented by massive sequencing techniques, which enable sequencing of very large DNA fragments. In addition to enabling study of the whole genome, they enable selective study of the exome or of a few selected genes. This makes it possible to detect genetic changes that are not even described in a specific tumor. Next-generation sequencing combines sequencing of huge amounts of DNA at a cost that can be assumed in daily diagnostic practice. The technique can reveal numerical changes, mutations, deletions, insertions, and rearrangements and has been used successfully in the classification and diagnosis of melanoma.77
ConclusionsRecent evidence shows that malignant melanocytic tumors develop along pathways involving cumulative genetic and epigenetic alterations and range from benign precursors, tumors of intermediate malignancy and malignant forms in situ right up to the malignant end of the spectrum. To date, 9 pathogenic pathways leading to melanoma have been identified. Some of these pathways are closely associated with exposure to sunlight, whereas others are not. Each pathway reveals identifiable characteristic genetic alterations that have proven essential for the correct diagnosis of the individual melanocytic tumor. Nevertheless, histopathology with hematoxylin–eosin staining continues to play a key role and constitutes the setting in which all other additional techniques should be considered.
Conflicts of InterestThe authors declare that they have no conflicts of interest.



