

Las proteínas de la vía RAS/MAPK (mitogen activated protein kinase pathway) desempeñan un papel fundamental en la proliferación, diferenciación, supervivencia y muerte celular. Desde hace más de 30 años se sabe que el 30% de los cánceres humanos presentan una mutación somática en alguno de los genes que codifican estas proteínas. En contraste con el elevado potencial de malignidad de las mutaciones somáticas, las mutaciones en la línea germinal provocan anomalías en el desarrollo del individuo que, si bien dependen específicamente del gen afectado, a menudo se superponen clínicamente. Así, todos los pacientes comparten un grado variable de retraso mental o dificultades de aprendizaje, trastornos cardiacos, dismorfismo facial, anomalías cutáneas y, en algunas instancias, predisposición al cáncer. Entre estos síndromes, conocidos como rasopatías, se incluyen el síndrome de Noonan, el síndrome de Costello, la neurofibromatosis 1, el síndrome LEOPARD, el síndrome cardio-facio-cutáneo y el síndrome de Legius. Es interesante conocer las manifestaciones cutáneas de las rasopatías, ya que estas pueden ayudar a esclarecer el diagnóstico de la enfermedad.
Proteins belonging to the RAS/mitogen activated protein kinase (MAPK) pathway play key roles in cell proliferation, differentiation, survival, and death. For more than 30years now we have known that 30% of human cancers carry somatic mutations in genes encoding proteins from this pathway. Whereas somatic mutations have a high malignant potential, germline mutations are linked to developmental abnormalities that are often poorly clinically differentiated, although each is dependent upon the specific gene affected. Thus, all patients share varying degrees of mental retardation or learning difficulties, heart disease, facial dysmorphism, skin anomalies, and, in some cases, predisposition to cancer. These syndromes, known as rasopathies, include Noonan syndrome, Costello syndrome, neurofibromatosis-1, LEOPARD syndrome, cardiofaciocutaneous syndrome, and Legius syndrome. Recognizing the skin manifestations of rasopathies can facilitate diagnosis of these syndromes.
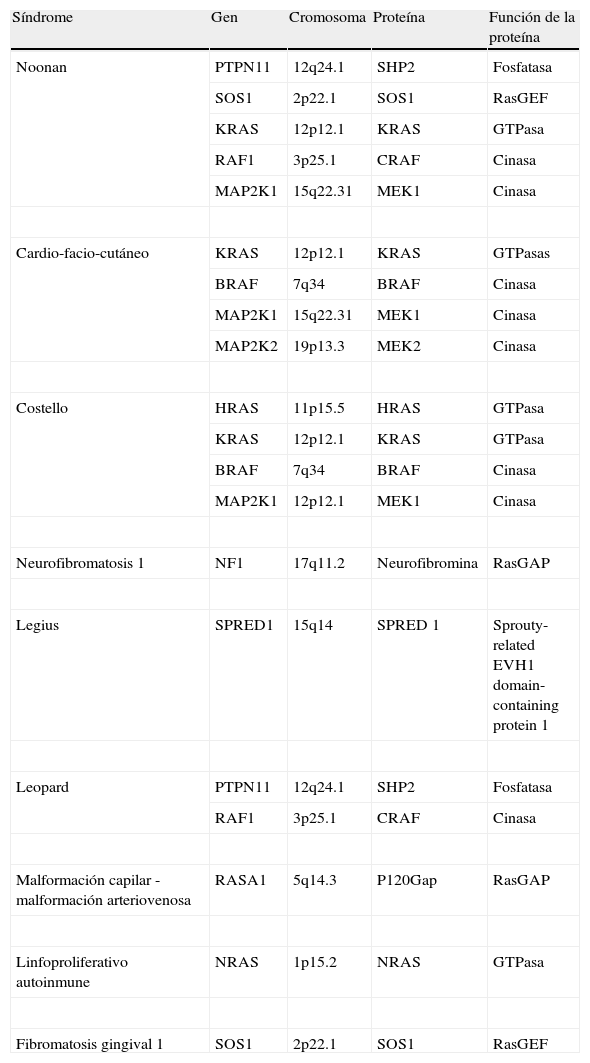
Los genes RAS desempeñan un papel esencial en la vía de señalización dependiente de la proteincinasa de activación mitogénica (MAPK), una cascada metabólica encargada de regular la proliferación, diferenciación, supervivencia y muerte celular. Estos genes deben su nombre a que fueron inicialmente identificados en muestras de tejido de cáncer de vejiga y pulmón como secuencias homólogas de los oncogenes de los virus de los sarcomas de las ratas v-Harvey (HRAS) y Kirsten (KRAS)1. Cada uno de los genes que codifican las proteínas de la vía RAS/MAPK se localiza en un cromosoma distinto y codifica una proteína diferente, por lo que su alteración provocará también una enfermedad diferente2 (tabla 1).
Caracterización biomolecular de las rasopatías
| Síndrome | Gen | Cromosoma | Proteína | Función de la proteína |
| Noonan | PTPN11 | 12q24.1 | SHP2 | Fosfatasa |
| SOS1 | 2p22.1 | SOS1 | RasGEF | |
| KRAS | 12p12.1 | KRAS | GTPasa | |
| RAF1 | 3p25.1 | CRAF | Cinasa | |
| MAP2K1 | 15q22.31 | MEK1 | Cinasa | |
| Cardio-facio-cutáneo | KRAS | 12p12.1 | KRAS | GTPasas |
| BRAF | 7q34 | BRAF | Cinasa | |
| MAP2K1 | 15q22.31 | MEK1 | Cinasa | |
| MAP2K2 | 19p13.3 | MEK2 | Cinasa | |
| Costello | HRAS | 11p15.5 | HRAS | GTPasa |
| KRAS | 12p12.1 | KRAS | GTPasa | |
| BRAF | 7q34 | BRAF | Cinasa | |
| MAP2K1 | 12p12.1 | MEK1 | Cinasa | |
| Neurofibromatosis 1 | NF1 | 17q11.2 | Neurofibromina | RasGAP |
| Legius | SPRED1 | 15q14 | SPRED 1 | Sprouty-related EVH1 domain-containing protein 1 |
| Leopard | PTPN11 | 12q24.1 | SHP2 | Fosfatasa |
| RAF1 | 3p25.1 | CRAF | Cinasa | |
| Malformación capilar -malformación arteriovenosa | RASA1 | 5q14.3 | P120Gap | RasGAP |
| Linfoproliferativo autoinmune | NRAS | 1p15.2 | NRAS | GTPasa |
| Fibromatosis gingival 1 | SOS1 | 2p22.1 | SOS1 | RasGEF |
Modificada de Tydiman et al2.
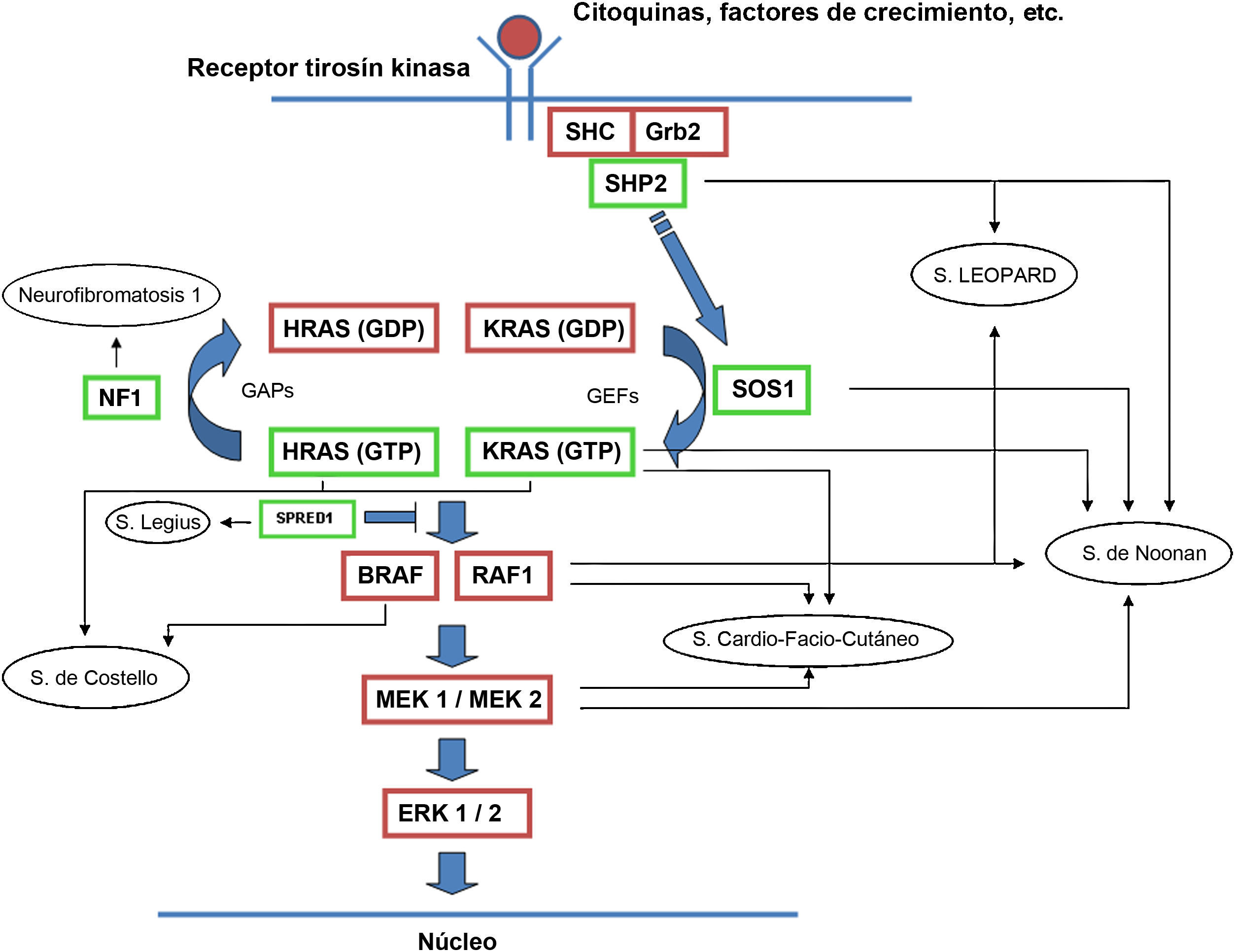
La familia de proteínas RAS es una subfamilia perteneciente a la superfamilia de proteínas G pequeñas, y a su vez consta de distintos componentes como las proteínas clásicas HRAS, KRAS, NRAS y otras como las R-RAS, TC21, M-RAS, Rap IA, Rap IB, Rap2A, Rap 2B, RaIA, and RaIB3. Las proteínas RAS actúan como mediadores esenciales en la transformación de estímulos extracelulares en señales intracelulares según su acoplamiento a GDP (forma inactiva) o GTP (forma activa)4,5. La estimulación de los receptores celulares por medio de citocinas, canales de calcio, integrinas, receptores de proteína G heteromérica, factores de crecimiento y otros péptidos, provoca la disociación del GDP de la proteína RAS y el posterior acoplamiento de la misma con el GTP, que la activa y promueve la interacción con diversos efectores como las proteínas RAF y MEK6,7. El cambio GDP/GTP es estimulado por factores de intercambio de nucleótido guanina (GEF) como el son-of-sevenless (SOS), el factor de liberación de guanina del RAS (RASGRF) y la proteína RAS de liberación del guanilo (RASGRP); cuando el receptor celular es estimulado, este se une a un dominio SH2 de las proteínas SHC, SHP2 y GRB2, las cuales reclutan al SOS intracitoplásmico que a continuación promueve el intercambio GDP/GTP en las proteínas RAS. Para limitar la activación de las proteínas RAS existe tanto una actividad GTPasa intrínseca como una actividad GTPasa estimulada por las proteínas activadoras de GTPasa (GAPs), que determinan el intercambio de la forma activa unida a GTP a la forma inactiva unida a GDP (fig. 1). La activación de RAS se acompaña de la activación de RAF (ARAF, BRAF, CRAF), que es la primera proteína MAPK de la vía metabólica; a continuación, se activa ERK1/ERK2, que son los últimos efectores de la vía con función sobre una gran cantidad de moléculas tanto citosólicas como intranucleares, últimas responsables del mantenimiento del ciclo de vida celular8.
 y limitado por proteínas GTPasa (GAPs) que determinan el paso a la forma inactiva unida a GDP. La activación de las proteínas RAS se acompaña de la activación de RAF (BRAF, RAF1), MEK1A1/MEK1A2 y, por último, ERK1/ERK2, que son los últimos efectores de la vía RAS/MAPK y responsables del mantenimiento del ciclo de vida celular.")
Vía metabólica RAS y principales síndromes genéticos asociados a su alteración. Tras la estimulación de los receptores celulares se activan proteínas intracelulares como la SHC, la GRB2 y la SHP2, que a su vez reclutan al SOS1 intracitoplásmico. El SOS promueve el intercambio GDP/GTP en las proteínas RAS, las cuales se activan por medio de la fosforilación. La proteína RAS acoplada al GTP promueve la interacción con otros efectores como las proteínas RAF y MEK. El cambio GDP/GTP es estimulado por factores de intercambio de nucleótido guanina (GEF) y limitado por proteínas GTPasa (GAPs) que determinan el paso a la forma inactiva unida a GDP. La activación de las proteínas RAS se acompaña de la activación de RAF (BRAF, RAF1), MEK1A1/MEK1A2 y, por último, ERK1/ERK2, que son los últimos efectores de la vía RAS/MAPK y responsables del mantenimiento del ciclo de vida celular.
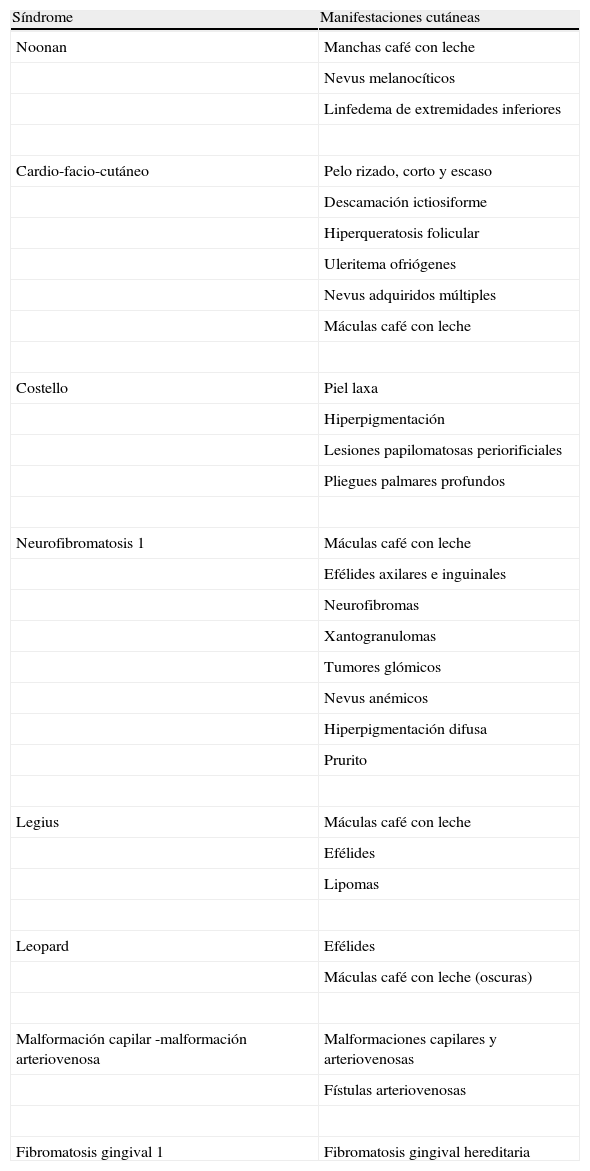
La vía RAS/MAPK fue inicialmente estudiada en el contexto de la oncogénesis, ya que su disregulación está presente en el 20-30% de los cánceres somáticos9. A diferencia de las mutaciones somáticas de la vía RAS, cuyo potencial de malignidad es muy elevado, las mutaciones en la línea germinal provocan anomalías en el desarrollo del individuo que, si bien dependen específicamente del gen afectado, a menudo se superponen clínicamente. Así, todos los pacientes afectados comparten un grado variable de retraso mental o dificultades de aprendizaje, trastornos cardiacos (fundamentalmente estenosis pulmonar y miocardiopatía hipertrófica), dimorfismo facial, macrocefalia, talla baja, anomalías cutáneas y, en algunas instancias, predisposición al cáncer (tabla 2). Probablemente, la superposición clínica se debe a que cada alteración repercute sobre el resto de los mediadores de la vía RAS/MAPK, ya que, a excepción del síndrome de LEOPARD, todas las mutaciones detectadas hasta el momento se caracterizan por un aumento de la actividad fisiológica de la proteína mutada y el consiguiente incremento de la señalización derivada de la misma10.
Manifestaciones cutáneas características de las rasopatías
| Síndrome | Manifestaciones cutáneas |
| Noonan | Manchas café con leche |
| Nevus melanocíticos | |
| Linfedema de extremidades inferiores | |
| Cardio-facio-cutáneo | Pelo rizado, corto y escaso |
| Descamación ictiosiforme | |
| Hiperqueratosis folicular | |
| Uleritema ofriógenes | |
| Nevus adquiridos múltiples | |
| Máculas café con leche | |
| Costello | Piel laxa |
| Hiperpigmentación | |
| Lesiones papilomatosas periorificiales | |
| Pliegues palmares profundos | |
| Neurofibromatosis 1 | Máculas café con leche |
| Efélides axilares e inguinales | |
| Neurofibromas | |
| Xantogranulomas | |
| Tumores glómicos | |
| Nevus anémicos | |
| Hiperpigmentación difusa | |
| Prurito | |
| Legius | Máculas café con leche |
| Efélides | |
| Lipomas | |
| Leopard | Efélides |
| Máculas café con leche (oscuras) | |
| Malformación capilar -malformación arteriovenosa | Malformaciones capilares y arteriovenosas |
| Fístulas arteriovenosas | |
| Fibromatosis gingival 1 | Fibromatosis gingival hereditaria |
Aunque la relación geno-fenotípica es incierta, algunos autores proponen dividir estos síndromes neuro-cardio-facio-cutáneos en tres grupos, según el lugar de afectación de la vía metabólica11: a) síndromes causados por alteración de los genes en la parte superior de la vía (upstream), es decir, PTPN11, SOS1 y neurofibromina. En este grupo los pacientes suelen tener un fenotipo de tipo Noonan, retraso mental leve y más tendencia a sufrir lesiones pigmentadas que manifestaciones ectodérmicas. Además, en el caso de la neurofibromina y el PTPN11, parece haber un riesgo ligeramente aumentado de sufrir leucemia; b) síndromes causados por la afectación del gen KRAS y los genes de la parte inferior de la cascada (downstream). En este caso se afectan principalmente las funciones cognitivas, el desarrollo general y el tegumento cutáneo, que muestra pliegues redundantes, trastornos de la queratinización y anomalías del pelo. Aunque el riesgo de malignidad asociada es bajo, pudiera incluir leucemias; y c) síndromes causados exclusivamente por las mutaciones en el gen HRAS, cuyo máximo exponente es el síndrome de Costello. En estos pacientes destacan la propensión a la propensión a fibrilación auricular, la hiperpigmentación cutánea, los crecimientos cutáneos papilomatosos y la tendencia a los tumores de partes blandas.
Adicionalmente, existen otras enfermedades relacionadas con mutaciones germinales en la vía RAS/MAPK como el síndrome linfoproliferativo autoinmune (ALPS)12, el síndrome de malformaciones capilares y malformaciones arteriovenosas (CM-AVM)13 o la hiperplasia fibrosa gingival tipo 1 (HFG1)14, que no conllevan defectos generalizados del desarrollo, pero que afectan a los mecanismos inmunológicos y la formación de los vasos sanguíneos, poniendo una vez más de manifiesto la importancia de la vía RAS/MAPK en la biología humana.
Manifestaciones cutáneas de las rasopatíasLas manifestaciones cutáneas no son específicas de cada síndrome. Los hallazgos dermatológicos más frecuentes en cada rasopatía se detallan en la tabla 3. De modo genérico, pueden dividirse en lesiones pigmentadas (máculas café con leche, lentigos, lesiones melanocíticas), lesiones ectodérmicas (manifestaciones ictiosiformes, hiperqueratosis folicular, pelo corto, rizado y escaso) y lesiones hiperplásicas (piel redundante, crecimientos papilomatosos).
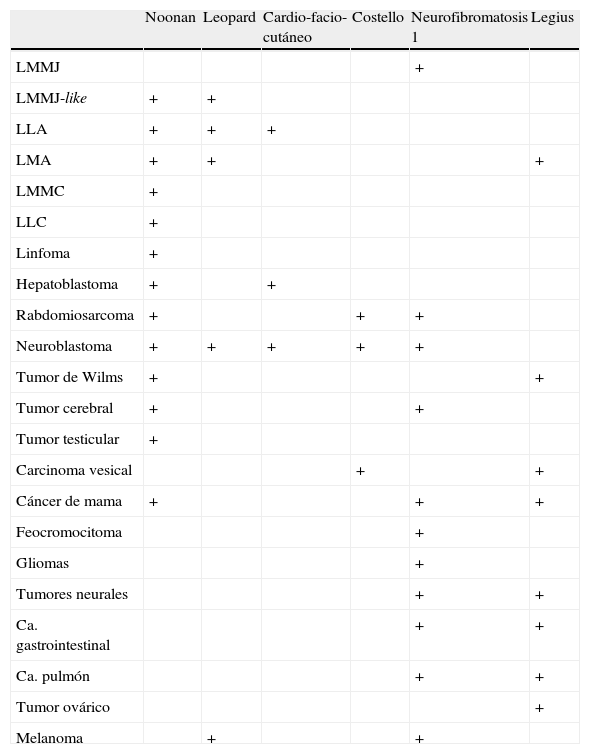
Enfermedades malignas descritas en rasopatías
| Noonan | Leopard | Cardio-facio-cutáneo | Costello | Neurofibromatosis 1 | Legius | |
| LMMJ | + | |||||
| LMMJ-like | + | + | ||||
| LLA | + | + | + | |||
| LMA | + | + | + | |||
| LMMC | + | |||||
| LLC | + | |||||
| Linfoma | + | |||||
| Hepatoblastoma | + | + | ||||
| Rabdomiosarcoma | + | + | + | |||
| Neuroblastoma | + | + | + | + | + | |
| Tumor de Wilms | + | + | ||||
| Tumor cerebral | + | + | ||||
| Tumor testicular | + | |||||
| Carcinoma vesical | + | + | ||||
| Cáncer de mama | + | + | + | |||
| Feocromocitoma | + | |||||
| Gliomas | + | |||||
| Tumores neurales | + | + | ||||
| Ca. gastrointestinal | + | + | ||||
| Ca. pulmón | + | + | ||||
| Tumor ovárico | + | |||||
| Melanoma | + | + |
Modificada de Hasle33. LLA: leucemia linfoblástica aguda; LLC: leucemia linfoide crónica; LMA: leucemia mieloide aguda; LMMC: leucemia mielomonocítica crónica; LMMJ: leucemia mielomonocítica juvenil; LMMJ-like: leucemia mielomonocítica juvenil-like.
Todavía no está esclarecido el papel de la vía RAS/MAPK en el desarrollo del tegumento cutáneo, así que se desconoce el mecanismo por el que aparecen las anomalías cutáneas. En un reciente estudio experimental se ha podido comprobar que la hiperactivación del KRAS bloquea la proliferación del pelo e induce la aparición de piel redundante, crecimientos papilomatosos y uñas cortas15.
Síndrome de NoonanEl síndrome de Noonan (SN, OMIM 163950) es un trastorno autosómico recesivo genéticamente heterogéneo. Hasta el momento se han descrito mutaciones en los genes PTPN11, SOS1, KRAS, RAF1 y BRAF16,17, pero es muy posible que haya otros involucrados. Aproximadamente la mitad de los casos sufren una mutación del gen PTPN1, un gen de 16 exones localizado en el cromosoma 12q24.118. El PTPN11 codifica una proteína tirosina-fosfatasa denominada SHP2, cuya actividad catalítica es autoinhibida mediante la interacción de dos de sus propios dominios, el N-terminal src homólogo 2 y el catalítico tirosín fosfatasa (PTP). Esta interacción se altera en la mayoría de las mutaciones que afectan al gen PTPN11, por lo que la actividad catalítica no se limita y la señalización de la vía RAS/MAPK resulta sobreestimulada19. Por otro lado, un número significativo de enfermos con síndrome LEOPARD también presenta mutaciones en el gen PTPN1120, lo cual justifica la similitud clínica entre ambos síndromes. La segunda causa más frecuente de aparición del SN (aproximadamente un 17-28% de los casos) es una mutación en el gen SOS121,22. El gen SOS1 codifica una proteína denominada SOS1 con actividad GEF, que estimula la conversión de la forma inactiva de Ras ligada a GDP a la forma activa ligada a GTP. Las mutaciones provocan un aumento de la función de la proteína SOS1, con la consiguiente hiperestimulación de la vía metabólica. Este mismo efecto aparece cuando las mutaciones, mucho más infrecuentes, se producen en los genes KRAS y RAF123 y BRAF17. Finalmente, recientemente se han descrito casos de SN con mutaciones en el gen NRAS24, y otros con fenotipo parecido al SN y alteración en el gen SHOC2, el cual codifica una proteína que participa en la unión de RAS con los efectores inferiores de la vía25 y en el CBL26, un gen supresor tumoral que se altera en las leucemias mieloides.
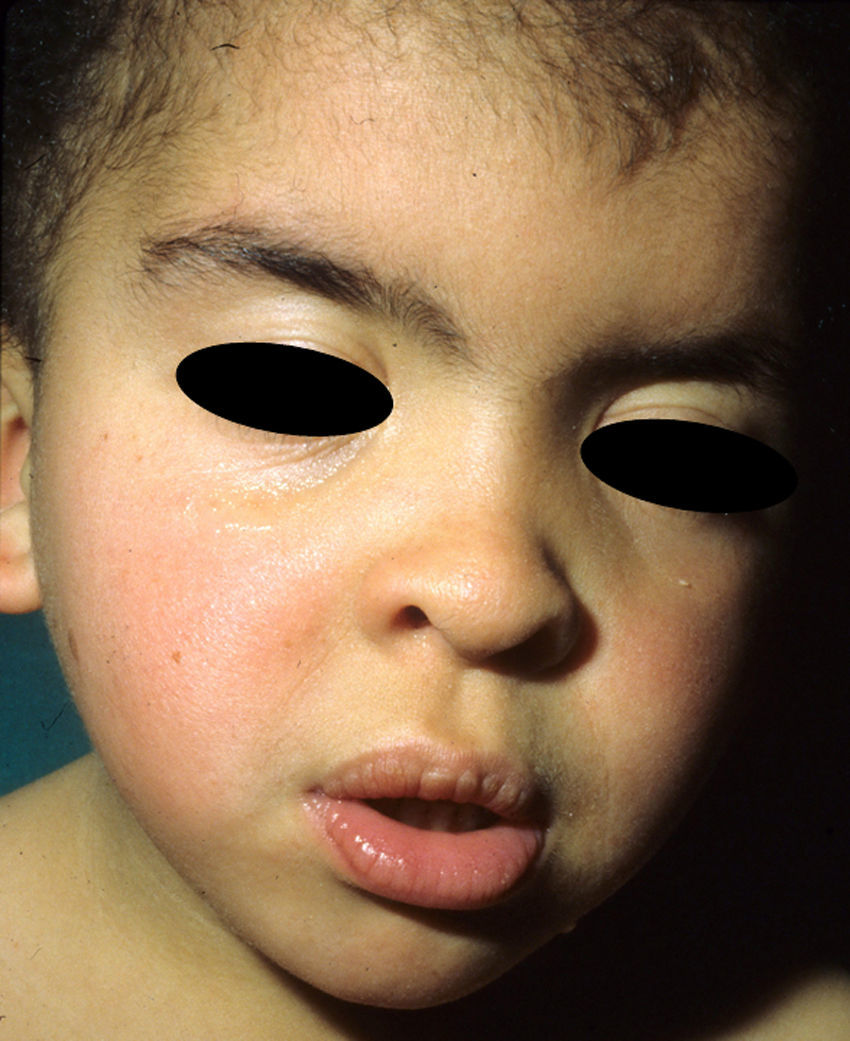
Los rasgos fenotípicos esenciales del SN incluyen anomalías faciales, talla baja, anomalías cardiacas congénitas, trastornos de coagulación y un grado variable de retraso cognitivo. En la cara se puede observar frente ancha, hipertelorismo, ptosis, pliegue epicántico, filtro labial alto y con los rebordes labiales con la angulación acentuada hacia arriba (fig. 2), orejas de implantación baja y rotadas posteriormente, cejas arqueadas o con un ángulo de vértice superior, iris azul claro, cuello ancho, y línea posterior de implantación del pelo baja. En el tórax es frecuente el pectus excavatum inferior/carinatum superior y las areolas separadas; en el 50-80% de los casos hay anomalías congénitas cardiacas, fundamentalmente estenosis pulmonar (20-50%) y cardiomiopatía hipertrófica (20-30%), pero también defectos del septo auricular y ventricular y tetralogía de Fallot. Menos frecuentemente se pueden observar criptorquidia y diátesis hemorrágica. El retraso mental es inconstante (15-35% de los pacientes) y no suele ser severo27,28. Aunque no hay una estricta correlación geno-fenotípica, las mutaciones en el gen PTPN11 se asocian significativamente con la presencia de estenosis valvular pulmonar, talla baja, diátesis hemorrágica y deformidades torácicas, mientras que los pacientes que no las presentan suelen tener alteraciones cardiacas y una cara menos característica29. Los pacientes con mutaciones en el gen SOS1 presentan estenosis pulmonar y llamativas anomalías ectodérmicas, pero los defectos septales atriales son menos frecuentes que en el caso de mutaciones en el PTPN1121,22. El fenotipo asociado con las mutaciones en el gen KRAS es menos definido, y a menudo se superpone con el del síndrome de Costello y el síndrome cardio-facio-cutáneo. Finalmente, las mutaciones en el gen RAF1 se asocian muy significativamente con la presencia de miocardiopatía hipertrófica (76% de prevalencia en este grupo frente al 18% en el resto de los pacientes con SN) y de lesiones cutáneas pigmentadas23, mientras que las mutaciones en BRAF parecen asociarse a un fenotipo más severo y a la presencia de lentigos oscuros y nevus melanocíticos múltiples17. En definitiva, en el SN hay tanta heterogeneidad clínica y genética que hace falta estudiar una serie de pacientes mucho más extensa para poder concretar la correlación geno-fenotípica.

Los pacientes con SN presentan discretas anomalías cutáneas, que parecen depender de la mutación causal. Así, las alteraciones ectodérmicas (pelo corto y rizado, alopecia de cejas, eritema cutáneo) son mucho más frecuentes en los casos con mutación en el gen SOS121,22, mientras que las mutaciones en el gen SHOC2 se asocian característicamente con el cabello anágeno suelto24. Las lesiones pigmentadas son más frecuentes en las mutaciones del gen RAF123 y del BRAF17 (fig. 3). Otras manifestaciones cutáneas del SN incluyen anomalías linfáticas30, tumores de células granulares31 y malformaciones vasculares capilares32.
Síndrome de Noonan y riesgo de cáncer.")
Los pacientes con SN tienen un riesgo elevado de sufrir malignidades hematológicas y otros tumores como rabdomiosarcoma, neuroblastoma, tumores de células gigantes o tumores testiculares, si bien el riesgo absoluto de padecerlos es relativamente bajo33. Los recién nacidos con mutaciones en el gen PTPN11 pueden sufrir un síndrome mieloproliferativo muy parecido a la leucemia mielomonocítica juvenil (LMMJ) que regresa espontáneamente33. Al contrario de lo que ocurre con el PTPN11, que actúa como un oncogén en distintos tumores, el gen SOS1 no aparece mutado en los cánceres humanos, por lo que para algunos autores los pacientes con una mutación en SOS1 no tendrían riesgo aumentado de sufrir una malignidad34. Algo similar ocurre con las mutaciones en RAF1, que parecen condicionar las anomalías cardiacas, pero que no han sido detectadas en cánceres somáticos.
Síndrome LEOPARDEl síndrome LEOPARD (SL, OMIM 151100) es un trastorno de herencia autosómica dominante cuyo nombre es un acrónimo que agrupa las principales manifestaciones clínicas: lentigos, anomalías electrocardiográficas, hipertelorismo ocular, estenosis pulmonar, anomalías genitales, retraso del crecimiento y sordera neurosensorial (Deafness en inglés). Se debe a mutaciones en el dominio PTP del gen PTPN11 que, al contrario de lo que ocurre en el SN, determinan una pérdida de función en la actividad catalítica del SHP-2. Como ya hemos comentado anteriormente, el fenotipo de los pacientes con SL y SN puede ser muy similar, pero se desconoce cómo es posible que mutaciones activadoras e inactivadoras de un mismo gen tengan una manifestación fenotípica similar. De hecho, también se han descrito pacientes con SL con mutaciones activadoras del RAF123, por lo que es probable que haya otros factores asociados que determinen la aparición del SN.
Los pacientes afectados presentan un fenotipo muy parecido al del SN, el cual se va acentuando con el paso de los años. Las lesiones lentiginosas pueden tardar en aparecer, dificultando el diagnóstico diferencial con el SN al principio. Las alteraciones del ECG están presentes en un 75% de los pacientes, incluyendo trastornos de la conducción, de la repolarización y del eje QRS35. Casi todos los adultos tienen hipertelorismo, además de puente nasal ancho, ptosis palpebral, orejas de implantación baja, pliegues nasolabiales acentuados y arrugas prematuras en la cara36. La estenosis valvular pulmonar se creía presente en el 40% de los pacientes, pero actualmente se considera una frecuencia mucho menor, en torno al 10-20% de los casos35; por el contrario, la miocardiopatía hipertrófica progresiva es el defecto cardiaco más prevalente y es causa de muerte en no pocos casos36. La mitad de los pacientes tiene criptorquidia bilateral, y tampoco es raro encontrar hipospadias, hipoplasia genital y retraso de la pubertad37. El 25% de los pacientes adultos presenta una talla más baja de lo normal. La sordera neurosensorial afecta al 15-25% de los pacientes, y aunque suele ser diagnosticada en la infancia, también puede aparecer tardíamente36. El retraso mental, si existe, es leve.
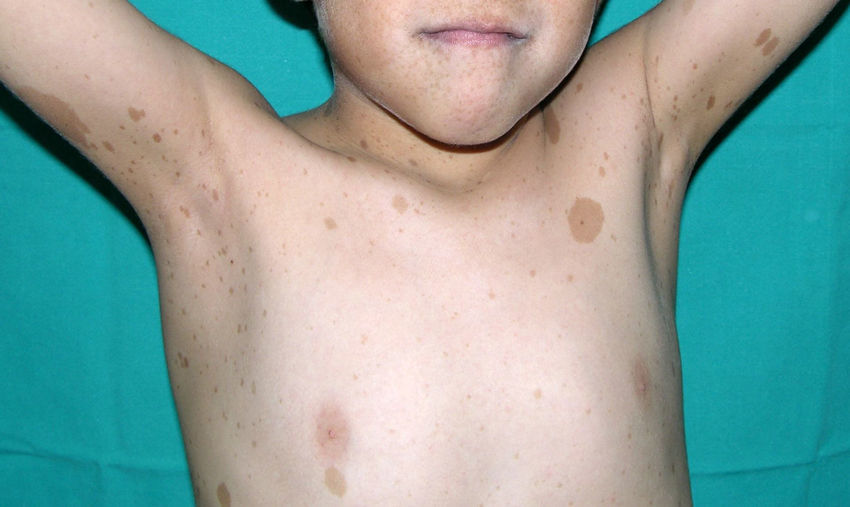
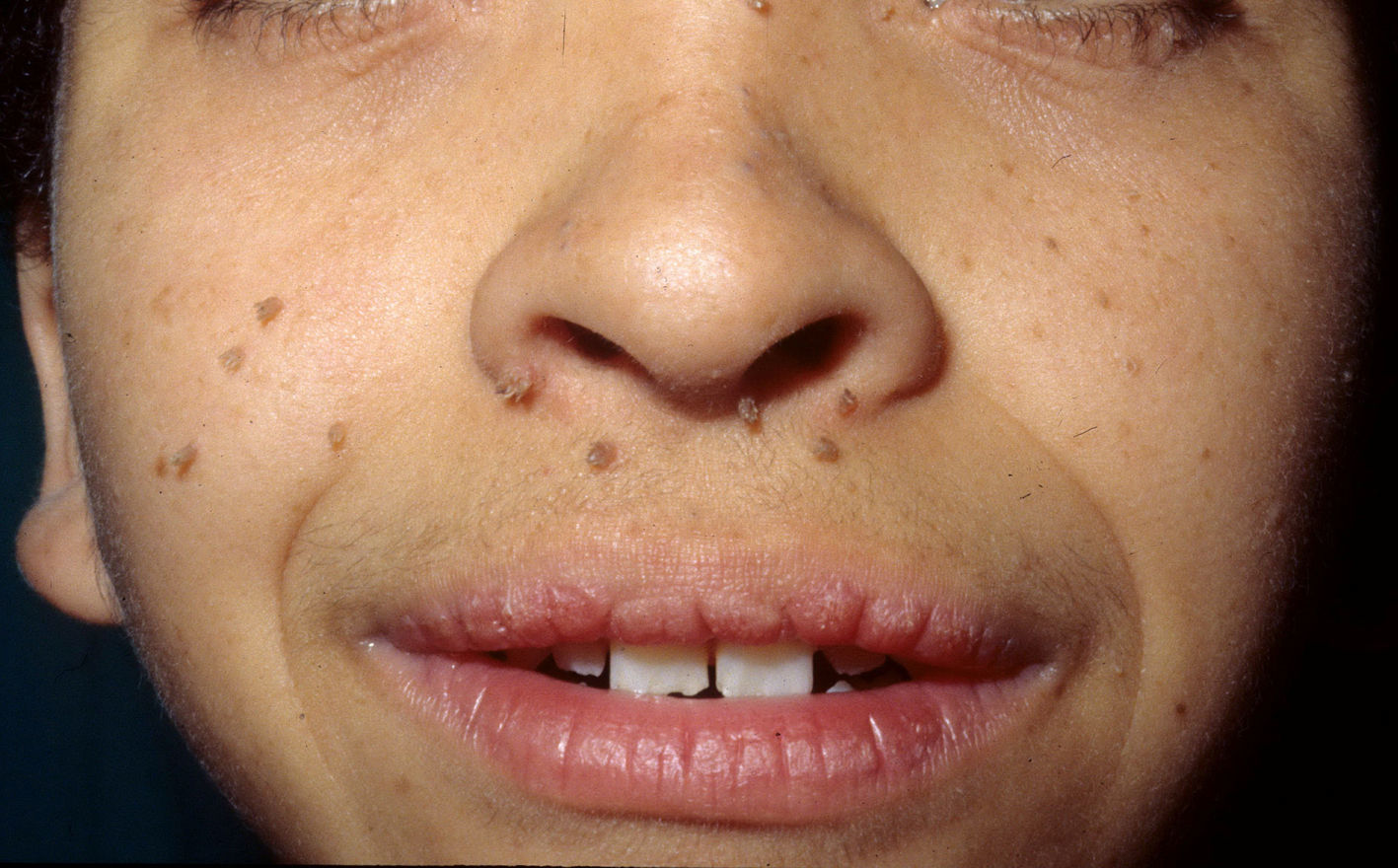
Manifestaciones cutáneasLas lesiones lentiginosas son el hallazgo cutáneo más característico, pero pueden no estar presentes en los primeros años de la vida38,36, dificultando el diagnóstico diferencial con el SN. Las lesiones son de tono marrón oscuro y afectan a todo el cuerpo, pero son particularmente llamativas en la cara y la parte superior del cuerpo; aparecen a partir de los 4-5 años, y su número aumenta exponencialmente a lo largo de la infancia, respetando siempre las mucosas, independientemente de la exposición solar (fig. 4). En un 50% de los pacientes también se observan máculas café con leche (MCCL) típicas o de tono mucho más oscuro de lo habitual (en ese caso denominadas máculas café noir o café negro), pudiendo estar presentes incluso antes que las lesiones lentiginosas36,38. La histología de las máculas más oscuras puede corresponder a lentigos simples o a nevus melanocíticos, por lo que algunos autores proponen incluir las lesiones melanocíticas en el espectro del síndrome LEOPARD39. Más raramente puede haber lesiones hipopigmentadas, y se han comunicado casos de melanoma40.
.")
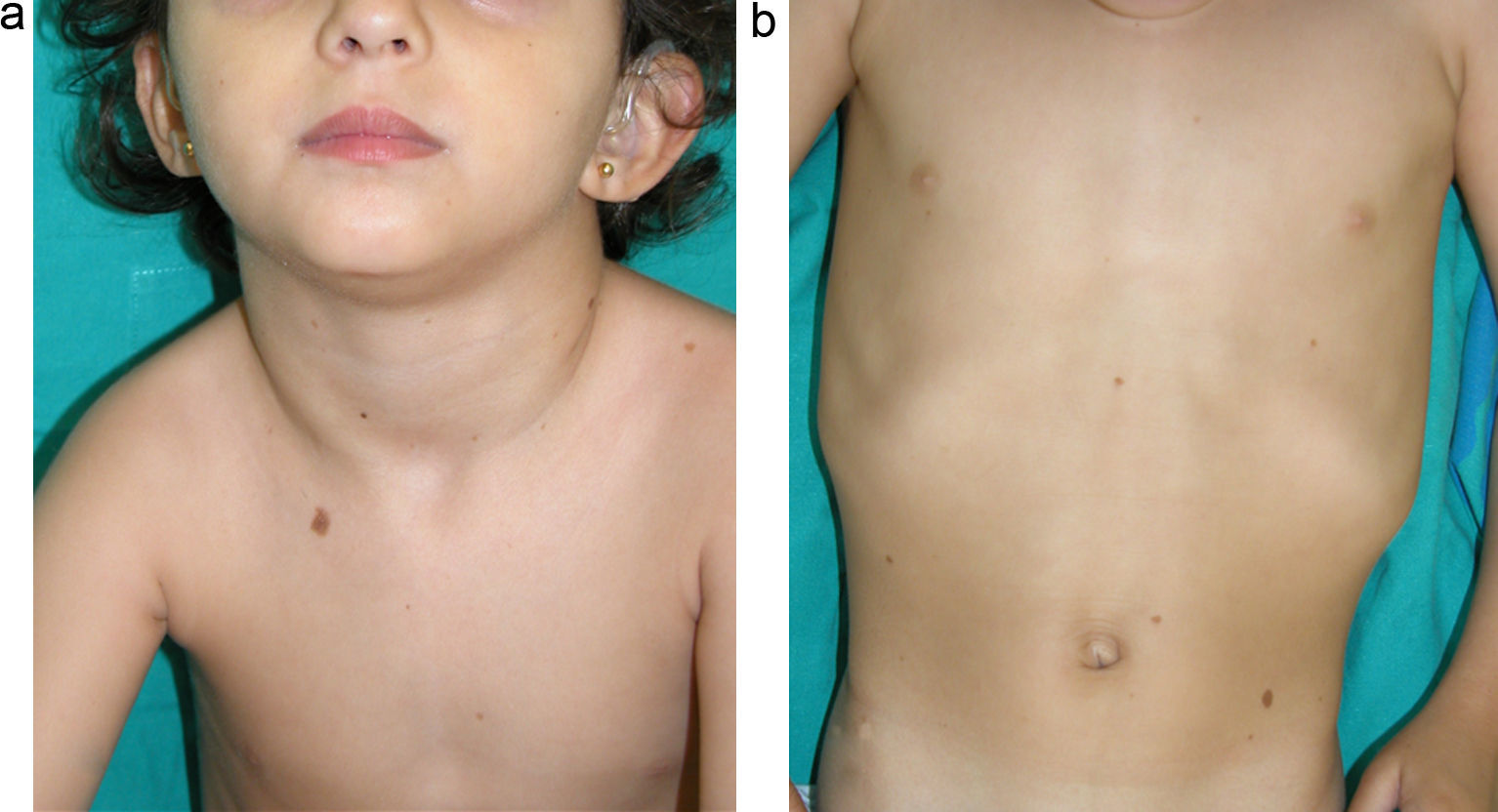
Síndrome de LEOPARD. a. Lentigines en la parte superior del tronco de una niña con síndrome de LEOPARD. Esta paciente, en la que se demostró una mutación en el gen PTPN11, presentaba sordera e hipertelorismo. Obsérvense también las orejas de implantación baja y el cuello ancho, hallazgos fenotípicos frecuentes en el síndrome de Noonan. b. Lentigines en cara anterior del tronco (Cortesía de la Dra. Ana Martín Santiago).
Se han descrito malignidades hematológicas como leucemia mieloide aguda, leucemia linfoblástica aguda y trastornos mieloproliferativos, así como neuroblastoma33, coristomas bilaterales y el ya mencionado melanoma40.
Neurofibromatosis tipo 1La neurofibromatosis tipo 1 (NF1, OMIM 162200) es un trastorno neurocutáneo de herencia autosómica dominante en el que en el 50% de los casos aparece de novo. Se debe a una alteración del gen de la neurofibromina, un gen con 61 exones localizado en el cromosoma 17q11.2 que tiene el índice de mutaciones espontáneas más alto de todo el genoma41. La neurofibromina es una proteína de 327kDa que ejerce un efecto regulador negativo en la vía RAS/MAPK. Contiene un dominio central con una proteína con capacidad activadora de GTPasa (GAP), lo cual se traduce en el paso de la proteína RAS a la forma inactiva ligada a GDP. La disminución de su actividad supresora se traduce en una actividad incrementada de toda la vía RAS/MAPK. La mayoría de los pacientes con NF presentan una mutación intragénica en la neurofibromina, y sólo un 5% de los mismos tiene una microdeleción de 1,4 megabases que contiene al menos 11 genes42. No existe correlación geno-fenotípica, salvo en dos tipos de anomalía genética: a) los pacientes con esta deleción suelen ser más altos, tener un mayor número de neurofibromas y presentar más dificultades de aprendizaje, un fenotipo facial más pronunciado y mayor propensión a los tumores malignos de la vaina nerviosa periférica que aquellos con una mutación intragénica43–45, y b) aquellos que tienen una deleción de tres pares de bases en el exón 17, que presentan ausencia de neurofibromas y una menor incidencia de complicaciones graves46.
La expresividad de las manifestaciones clínicas en la NF1 es muy variable, incluso dentro de una misma familia, y son dependientes de la edad47. En los años 80 se determinaron una serie de criterios diagnósticos que incluyen los siguientes hallazgos: a) 6 o más manchas café con leche (MCCL) de diámetro mayor de 5mm de diámetro máximo en la etapa prepuberal y mayor de 15mm en la etapa pospuberal; b) dos o más neurofibromas (NF) de cualquier tipo, o un neurofibroma plexiforme; c) pecas axilares o inguinales; d) dos o más nódulos de Lisch (hamartomas del iris); e) glioma óptico; f) una lesión ósea característica como displasia del esfenoides o adelgazamiento de la cortical ósea de los huesos largos con/sin pseudoartrosis; y g) un familiar en primer grado (hijo, hermano, padre o madre) con los criterios anteriores. Estos criterios son bastante sensibles y específicos en adultos, pero menos sensibles en niños por debajo de 8 años48. Además, no tienen en cuenta otros síntomas como la dificultad de aprendizaje, los tumores malignos de la vaina nerviosa o la macrocefalia. Un estudio retrospectivo publicado en el año 2000 que recogió 1.900 casos de NF1 encontró que el 46% de los casos esporádicos no cumplían criterios diagnósticos antes del primer año de vida, mientras que a los 8 años los cumplían el 97% y el 100% de los pacientes a los 20 años49.
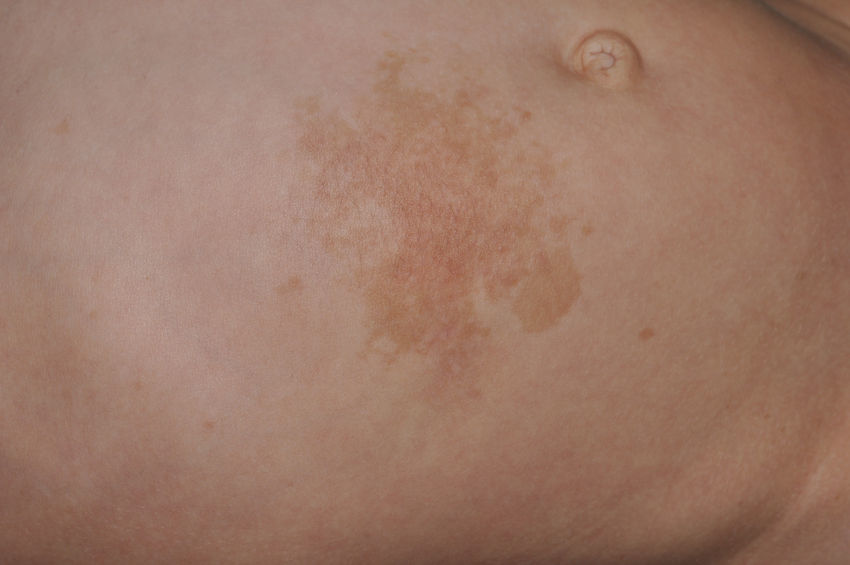
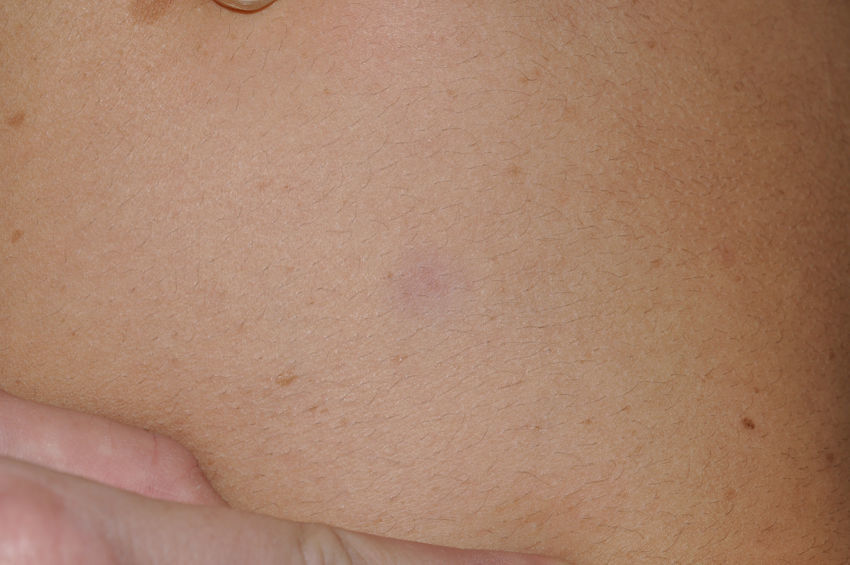
Manifestaciones cutáneasLos hallazgos dermatológicos más típicos forman parte de los criterios diagnósticos detallados más arriba (MCCL, pecas axilares e inguinales y NF). Las MCCL aparecen a lo largo del primer año de vida en el 99% de los pacientes, y suelen aumentar en número durante la infancia. Aunque pueden existir en pacientes sanos sin NF1 asociada, se calcula que menos del 1% de los niños menores de 5 años sanos tienen más de 2 MCCL50. Las MCCL respetan palmas, plantas y cuero cabelludo y son de color y tamaño variable, incluso dentro de un mismo paciente51. En general, tienden a aclararse a lo largo de los años y no suelen producir problemas estéticos. Las pecas axilares e inguinales (clásicamente conocidas como signo de Crowe) se observan entre el tercer y el quinto año de la vida; se consideran el hallazgo cutáneo más específico, para algunos autores casi patognomónico48. Casi el 90% de los adultos tienen pecas, y a menudo no sólo se limitan a los pliegues, sino que se extienden a lo largo del tronco, el cuello e incluso en torno a los labios52 (fig. 5). Los NF pueden aparecer en cualquier parte del cuerpo, pero lo hacen más tardíamente, generalmente después de la pubertad. Su tamaño es variable, y a menudo son la causa más importante de morbilidad debido a su volumen o visibilidad. Un subtipo especial es el NF plexiforme superficial, que suele ser congénito; los NF plexiformes superficiales presentan hiperpigmentación e hipertricosis, por lo que a menudo se confunden con un nevus melanocítico congénito (fig. 6). Entre las variantes inusuales de NF1 se cuentan las máculas rojoazuladas y pseudoatróficas (fig. 7), lesiones con un aspecto peculiar en relación con la presencia de tejido neurofibromatoso infiltrando los vasos dérmicos o rodeándolos respectivamente53. Entre los hallazgos cutáneos no tan característicos, pero relativamente frecuentes de la NF1, se incluyen xantogranulomas juveniles, tumores glómicos, melanomas, nevus anémicos, la hiperpigmentación generalizada y el picor48.
, el tronco e incluso en la cara.")


Existe una forma de NF1 en mosaico denominada NF segmentaria que se caracteriza por la afectación de un segmento del cuerpo y la ausencia de antecedentes familiares de la enfermedad. Puede haber MCCL aisladas, neurofibromas aislados, MCCL y neurofibromas simultáneamente o neurofibromas plexiformes aislados54. Se desconoce el riesgo de transmisión de la enfermedad, pero este existe si coexiste con mosaicismo gonadal55. Además, se sabe que el riesgo no depende de la localización, por lo que la posibilidad de transmisión existe incluso cuando la forma segmentaria respeta la zona genital54.
Neurofibromatosis 1 y riesgo de cáncerTodos los pacientes con NF1 sufren una mayor predisposición al desarrollo de neoplasias, ya sean benignas (como los NF o los tumores glómicos) o malignas. Del 8 al 12% de los pacientes con NF1 desarrollarán un tumor maligno de la vaina nerviosa periférica, generalmente en la zona de un NF plexiforme56. El dolor persistente y el crecimiento acelerado deben considerarse signos de alarma, y el picor local podría indicar la presencia de un tumor del sistema nervioso central57. Por otro lado, hay mayor incidencia de sarcomas58, rabdomiosarcomas, neuroblastomas, tumores gastrointestinales estromales, feocromocitomas y cáncer de mama59. Finalmente, los niños con NF1 tienen un riesgo entre 200 y 500 veces mayor al de la población normal de sufrir una LMMJ, independientemente de la concurrencia o no de xantogranulomas juveniles60; sin embargo, no parece adecuado hacer despistaje rutinario de LMMJ en estos niños, salvo que los hallazgos asociados lo aconsejen (hepatoesplenomegalia, adenopatías, palidez, petequias, etc.).
Síndrome de LegiusEl síndrome de Legius (SLG, OMIM 611431) o NF1-like es un síndrome parecido a la NF1 que fue descrito en 200761. Esta enfermedad, de herencia autosómica dominante, se debe a una mutación del gen SPRED1 (sprouty related EVH1 domain-containing protein 1), un gen de 7 exones localizado en el cromosoma 15q13.2, cuya alteración determina una pérdida de su función represora sobre la proteína RAF62. Por consiguiente, se produce un aumento en los niveles de las proteínas RAF1, MEK y ERK, así como un incremento en el factor de transcripción ELK1. Se han detectado mutaciones en todos los exones del gen, algunas de ellas recurrentes, pero sin claros puntos mutacionales «calientes»63. El SLG se caracteriza por la presencia de MCCL y/o lentigos, a lo que ocasionalmente se asocia macrocefalia, un fenotipo tipo Noonan y/o dificultades de aprendizaje. A menudo, estos pacientes cumplen criterios de NF1, pero en el SLG se excluye específicamente la presencia de nódulos del Lisch, neurofibromas, tumores del SNC y mutaciones en el gen de la NF161.
Manifestaciones cutáneasLos hallazgos cutáneos típicos del SLG incluyen manchas café con leche y lentígines. Algunos adultos presentan lipomas62,63. A la vista de las series publicadas parece que la mayor probabilidad de encontrar pacientes con SLG se da en los casos familiares de MCCL con o sin lentigos63.
Síndrome de Legius y riesgo de cáncerNo está claro si en el SLG existe una mayor predisposición al cáncer; se han descrito pacientes con leucemia monoblástica aguda64, cáncer de pulmón, tumor de Wilms, adenocarcinoma de colon61, cáncer de mama, tumor dermoide de ovario65 y un schwannoma vestibular63, por lo que es necesario tener en cuenta esa posibilidad.
Síndrome de CostelloEl síndrome de Costello (SC, OMIM 218040) es un trastorno esporádico de probable herencia autosómica dominante cuyo gen causal se descubrió en el año 200566, constituyendo el primer trastorno genético identificado consecuencia de una mutación germinal en la vía RAS. Prácticamente la totalidad de los pacientes con SC presentan una mutación en el gen que codifica la proteína HRAS67. Este gen se localiza en el cromosoma 11p15.5, se extiende a lo largo de 6,5 Kb y consta de 6 exones4. Codifica dos proteínas distintas, la p21-HRAS y la p19-HRAS; la p21-HRAS es mucho más abundante que la 19p-HRAS, la cual parece ejercer un posible efecto regulador negativo sobre aquella68,69. La mutación del HRAS produce una activación aberrante de la vía RAS. Casi todos los pacientes presentan una mutación heterozigota en el cromosoma paterno, pero existen casos aislados en los que el cromosoma alterado procedía de la madre70,71, e incluso algunos casos excepcionales de mosaicismo72,73. En una serie de 139 casos se demostró que la mutación pG12S estaba presente hasta en un 80% de los casos, y la pG12A en otro 7% más74, por lo que los análisis genéticos deben iniciarse por el exón 2, que es donde se localiza la mutación en la gran mayoría de los casos. El resto de los pacientes presentaban otras mutaciones, nunca iguales en más de 4 de ellos. Las mutaciones más comunes originan un fenotipo típico, mientras que otras como la pT85I se asocian con un fenotipo más leve75 o la pG12V con un fenotipo más severo66,76. El hecho de que todos los pacientes presenten una mutación heterozigota apunta a un patrón hereditario autosómico dominante; todos los casos publicados hasta el momento ocurrieron de novo en los padres, cuya media de edad oscilaba entre los 38 y los 43 años77,78. También se han publicado pacientes con fenotipo de SC con una mutación en el gen KRAS79,80 y otro en el BRAF81, poniendo de manifiesto todavía más el amplio espectro fenotípico de los pacientes con mutaciones en los genes de la vía RAS.
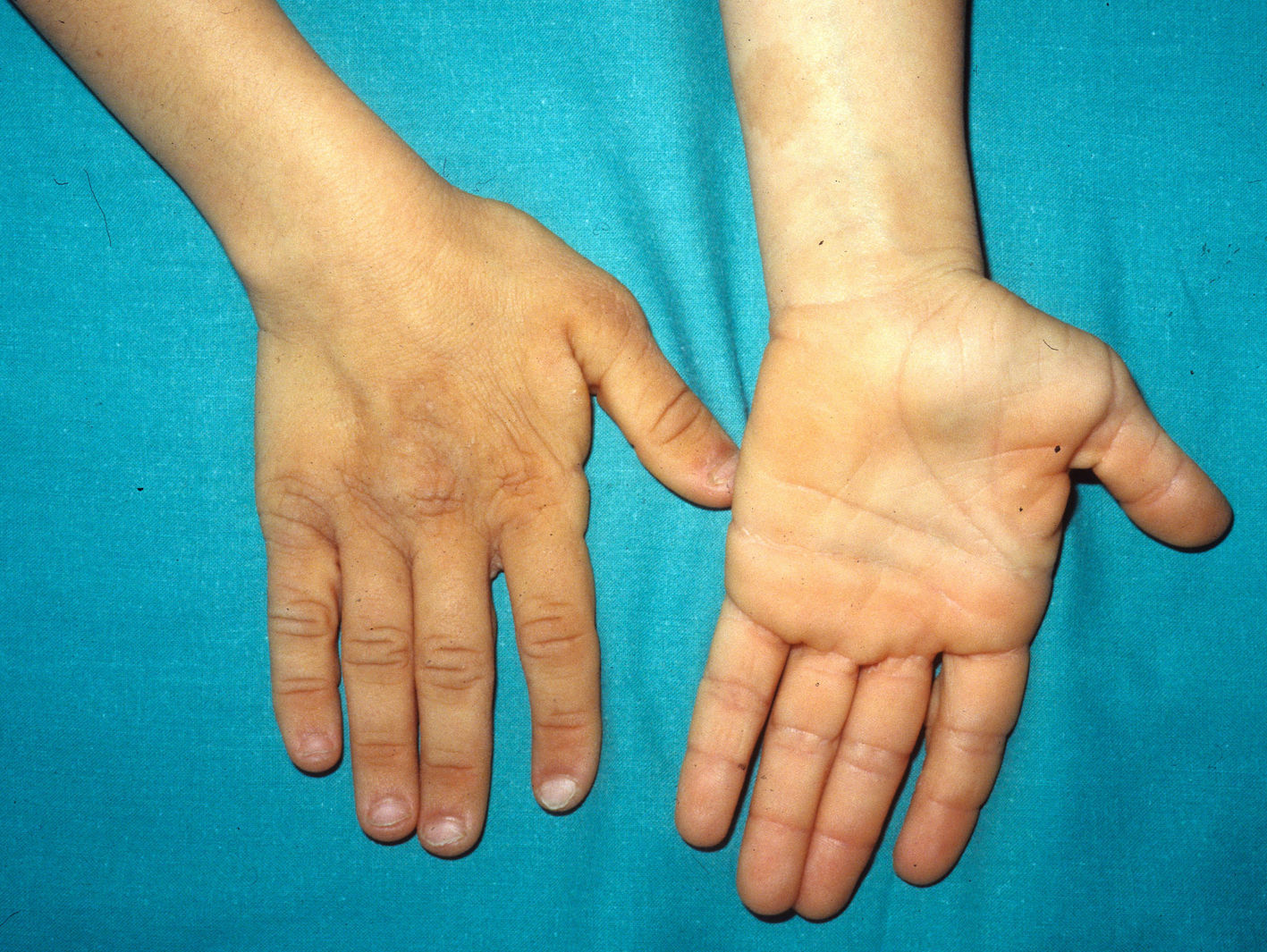
La gestación de estos pacientes cursa con polihidramnios (90%), avanzada edad parental (62%), macrosomía (50%), parto prematuro (50%) y, con menos frecuencia, taquiarritmia fetal74. En el periodo postnatal se observan dificultades importantes para la alimentación que suelen requerir la implantación de una sonda nasogástrica. Con el tiempo persiste un cierto retraso estatoponderal, y aunque el diámetro cefálico está dentro de los límites normales, el paciente tiene un aspecto macrocefálico. El retraso generalizado del desarrollo y la hipotonía se manifiestan en los primeros meses de la vida, y se reflejan en un déficit cognitivo moderado (IQ 56-69) cuando el niño es algo mayor, y un déficit particularmente evidente en la expresividad facial. Es muy llamativa la sociabilidad de los pacientes, que a menudo interaccionan con el médico haciendo preguntas o comentarios. Los rasgos faciales dismórficos se acentúan con el tiempo, e incluyen una boca grande de labios carnosos, mejillas rellenas, frente prominente, implantación baja del pelo, pliegue epicántico, nariz corta con puente nasal deprimido, y orejas de implantación baja, rotadas posteriormente, y con hélix y lóbulos engrosados (fig. 8)82. La desviación cubital de la muñeca y del quinto dedo de la mano son muy características, así como la retracción del talón de Aquiles (casi constante), la cifoescoliosis y la osteopenia. La deficiencia de la hormona del crecimiento es frecuente, así como la disregulación del desarrollo puberal. Al menos dos tercios de los pacientes presentan algún tipo de anomalía cardiaca, incluyendo hipertrofia cardiaca (41%), defectos cardiacos congénitos (21%) y taquicardia supraventricular (33%) entre los hallazgos más frecuentes83.

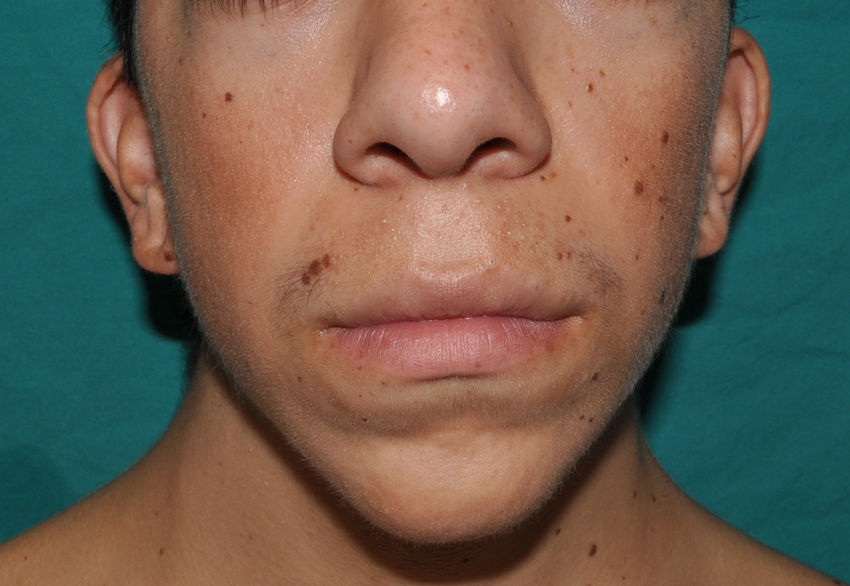
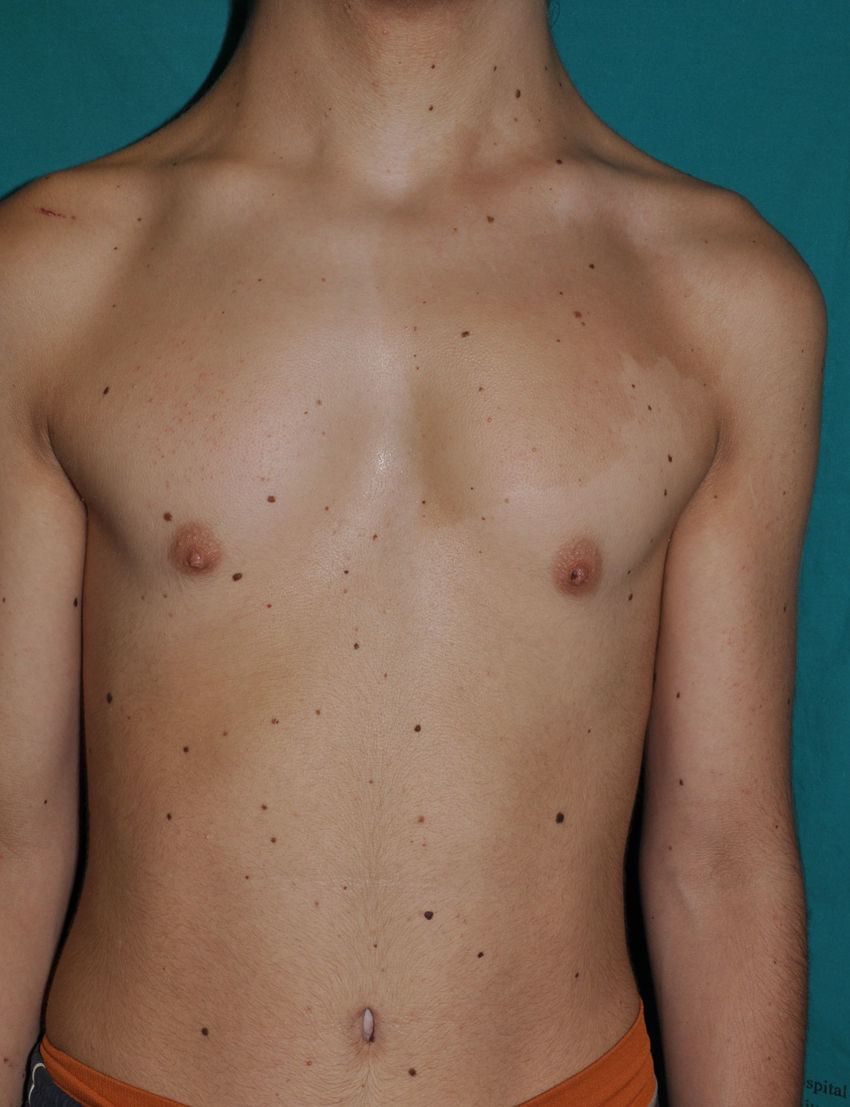
Los hallazgos cutáneos más significativos, aunque no patognomónicos, son el pelo rizado, escaso y corto, las lesiones papilomatosas periorificiales en la cara (fig. 9), la hiperpigmentación de los pliegues, la laxitud cutánea, la piel redundante en el dorso de las manos y los pies y la marcada acentuación de los pliegues palmoplantares (fig. 10). Las lesiones papilomatosas periorificiales pueden aparecer desde el primer año de vida o más tardíamente, durante la adolescencia82. El estudio histológico en estos pacientes revela un aumento de la fragmentación y anastomosis de las fibras elásticas en comparación con la piel normal84. Al parecer, los fibroblastos de estos pacientes presentan una deficiencia funcional de la proteína ligadora de elastina, que repercute en una alteración de las fibras elásticas y en un depósito anormal de las mismas en la piel, la lengua, la faringe, la laringe, los alvéolos y la aorta85,86.


Los pacientes con SC tienen una tendencia elevada al desarrollo de neoplasias, sean benignas o malignas. Un 13-15% de los pacientes presentará un cáncer a lo largo de la vida20,74, siendo el rabdomiosarcoma embrionario el tumor más común, seguido del neuroblastoma, el cáncer de células transicionales de vejiga y otros menos frecuentes, como el ganglioneuroblastoma, el adenocarcinoma de vejiga o el neurinoma del acústico87. El rabdomiosarcoma y el neuroblastoma aparecen durante la infancia, en general antes del cuarto año de la vida, mientras que el cáncer de vejiga es más frecuente en adultos, aunque puede aparecer en cualquier momento después de los 10 años a partir de la primera década de la vida87,88. Curiosamente, no se ha comunicado la aparición de leucemia en ningún paciente33. La evidencia del riesgo aumentado de malignidad hace aconsejable un protocolo de seguimiento en estos pacientes88. Parece que la probabilidad de cáncer es mayor en los pacientes con una mutación G12A que en la mutación G12S20.
Síndrome cardio-facio-cutáneoEl síndrome cardio-facio-cutáneo (SCFC, OMIM 115150) es un trastorno genético raro de aparición esporádica y herencia autosómica dominante. Hasta el momento se han descrito mutaciones en los genes BRAF, MAP1K1, MAP2K2 y KRAS. El gen BRAF se localiza en el cromosoma 7q34, consta de 18 exones y se extiende a lo largo de 190Mpb. Entre el 40 y el 78% de los pacientes con SCFC presentan mutaciones germinales en este gen89,90, que casi nunca está alterado en el resto de las rasopatías. La mutación más prevalente es la Q257R, que se localiza en el exón 6, seguida de otras en los exones 11,12,14 y 15. Aproximadamente un 25% de los pacientes presenta una mutación en los genes MAP2K1 o MAP2K219, los cuales se localizan en los cromosomas 15q22 y 19p13.3 respectivamente. Ambos contienen 11 exones y codifican las proteínas MEK1 y 2 respectivamente; estas proteínas son activadas por la proteína RAF y tienen capacidad para fosforilar las proteínas ERK1 y ERK2. El gen KRAS se localiza en el cromosoma 12p12.1, consta de 6 exones y genera dos isoformas de ARNm, una de las cuales (KRASA4a) se expresa en todos los tejidos del organismo, mientras que la otra (KRASA4b) no se expresa en el corazón91. Menos del 1% de los pacientes presentan una mutación en este gen, siendo la más frecuente la D153V; esta mutación concreta altera la generación de la isoforma KRASA4b, lo cual indica que esta isoforma participa en el desarrollo humano20. Para algunos autores no existe una clara relación geno-fenotípica92, mientras que para otros sí la hay; así, los pacientes con mutaciones KRAS tendrían una incidencia menor de manifestaciones cutáneas90, mientras que los afectados con una mutación MEK tendrían un desarrollo mental normal11. Se han descrito mutaciones germinales de KRAS en el SCFC, el SN, el SC y en algunos pacientes con fenotipo intermedio SN/SCFC y SC/SN, poniendo de manifiesto una vez más el solapamiento clínico entre estos pacientes93.
Los hallazgos fenotípicos a menudo se superponen con los de otras rasopatías, fundamentalmente con los del SN. Algunos enfermos tienen rasgos tan parecidos, que durante un tiempo el SCFC se consideraba una forma severa de SN; sin embargo, el importante retraso mental y las anomalías cutáneas ectodérmicas orientan clínicamente hacia el SCFC a estos pacientes. Adicionalmente, el análisis genético permite hacer el diagnóstico de certeza, ya que en el SCFC nunca hay mutaciones en el gen PTPN11. En los casos afectados suele haber polihidramnios durante la gestación y retraso del crecimiento en la etapa posnatal debido a las dificultades para la alimentación. Los rasgos dismórficos faciales incluyen una frente ancha, con constricción temporal bilateral, hendiduras palpebrales incurvadas hacia abajo, puente nasal deprimido y orejas con hélix prominentes y rotadas hacia atrás. El retraso psicomotor es de moderado a severo, y una o más anomalías cardiacas están presentes en el 75% de los pacientes, fundamentalmente estenosis pulmonar (45%), miocardiopatía hipertrófica (40%) y defectos del septo auricular (22%)93.
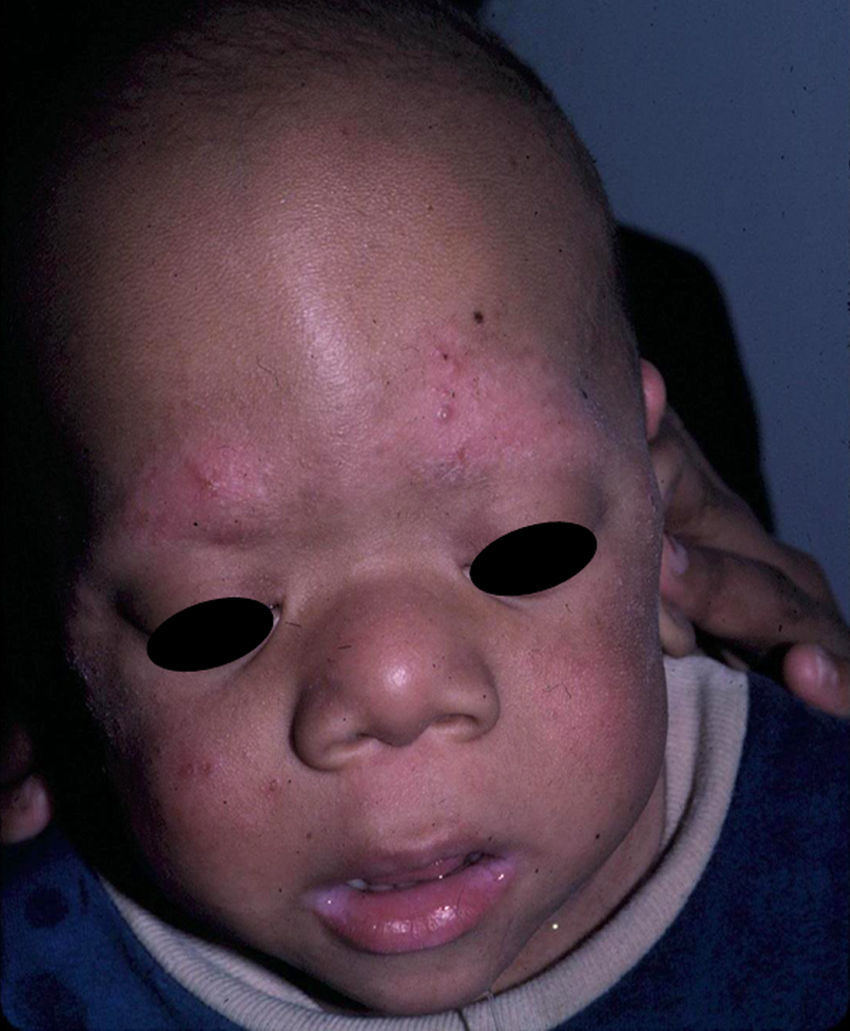
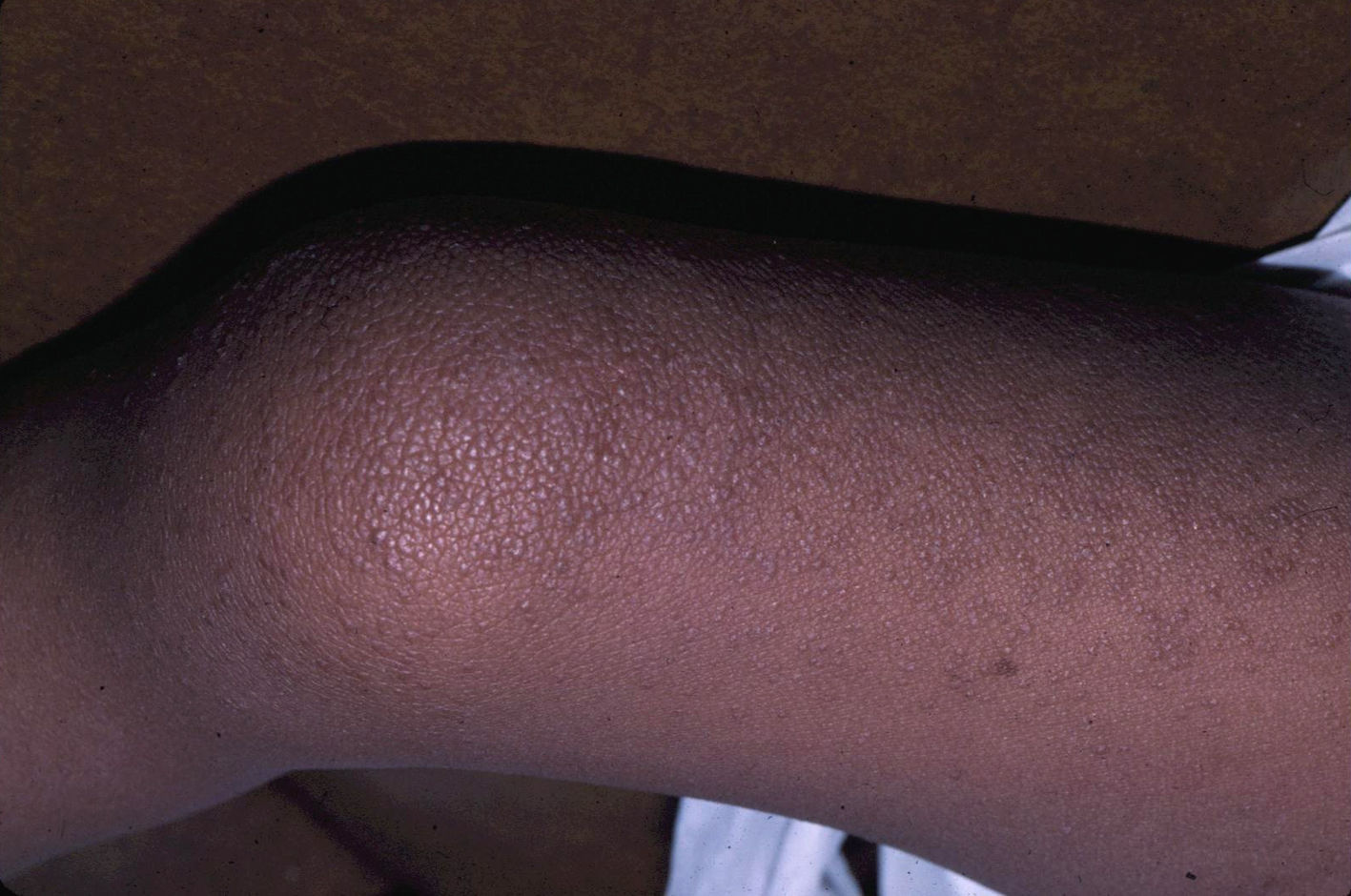
Manifestaciones cutáneasEn el SCFC las anomalías ectodérmicas son significativas, y a menudo permiten orientar el diagnóstico diferencial. Los pacientes tienen el pelo corto, escaso y rizado, descamación ictiosiforme e hiperqueratosis folicular generalizada (fig. 11). Un hallazgo distintivo es el uleritema ofriógenes, con hiperqueratosis folicular, eritema y alopecia cicatricial de las cejas (fig. 12). Cuando los individuos alcanzan la edad adulta puede aparecer hiperqueratosis palmoplantar y linfedema. En una serie de 61 pacientes, publicada en 2010, se observaron anomalías en el pelo en el 93% de los pacientes, uleritema ofriógenes en el 90% de los pacientes, queratosis pilar en el 80%, hiperqueratosis palmoplantar en el 36%, más de 50 nevus melanocíticos adquiridos en el 23% y hemangiomas infantiles en el 26% de los casos94. Además, se mencionan otros hallazgos cutáneos como la aceleración en la velocidad de crecimiento de las uñas, la escasez de vello en las extremidades, el olor axilar prepuberal, el escaso crecimiento del cabello, el pliegue de los lóbulos de las orejas, la acantosis nigricans o los pezones hiperplásicos. Aunque en esta serie no se observó ningún paciente con MCCL ni papilomas periorificiales, hallazgos comunes en otras rasopatías, otros autores sí han encontrado pacientes con MCCL y lentigos17,95. Finalmente, algunos autores apuntan la posible afectación de los ductos ecrinos y el folículo piloso, ya que en el estudio histológico de un paciente se observaron metaplasia escamosa ecrina y granulomas perianexiales96.
.")
.")
Al contrario de lo que ocurre en otros síndromes de la vía RAS/MAPK, en el SCFC no parece haber riesgo aumentado de malignidad. No obstante, se han descrito dos pacientes con una mutación germinal en BRAF que desarrollaron una leucemia linfoblástica aguda90,97,98, otro con hepatoblastoma metastásico99, y uno más con un neuroblastoma90, por lo que no se debe despreciar este riesgo.
Síndrome linfoproliferativo autoinmuneEl síndrome linfoproliferativo autoinmune (OMIM 164790) se caracteriza por un defecto en la apoptosis de los linfocitos que determina el cúmulo de linfocitos normales en el organismo y un riesgo aumentado de desarrollar malignidades hematológicas100. La mayoría de los casos se debe a un defecto de la apoptosis mediada por el receptor extrínseco Fas secundario a mutaciones en la ruta del CD95101, pero recientemente se han identificado mutaciones germinales en NRAS que producen esta misma enfermedad por un mecanismo independiente12. La mutación en NRAS produce una estabilización en la forma activa ligada a GTP, que resulta en una activación de toda la vía RAS. Así, aumenta la fosforilación de la proteína ERK, que inhibe la expresión linfocítica del BIN y, secundariamente, la apoptosis mitocrondrial intrínseca.
Síndrome de malformación capilar-malformación arteriovenosaEl síndrome de malformación capilar-malformación arteriovenosa (OMIM 608354) se trata de un trastorno autosómico dominante en el que malformaciones capilares multifocales se asocian a malformaciones vasculares y fístulas arteriovenosas en la piel, músculos, huesos y órganos internos como el cerebro y el corazón13,102. Se debe a mutaciones inactivadoras del gen RASA1 que, al igual que el gen NF1, codifica una proteína Ras-GAP; la insuficiencia de esta proteína conlleva una reducción en la hidrólisis de Ras-GTP y una sobreestimulación secundaria de la vía RAS/MAPK102. Se desconoce si los pacientes tienen un riesgo aumentado de sufrir tumores.
Fibromatosis gingival hereditariaLa fibromatosis gingival hereditaria (FGH, OMIM 135300) se caracteriza por el crecimiento fibroso benigno, lento y progresivo de las encías. Es un proceso genéticamente heterogéneo de herencia autosómica dominante o recesiva según cada caso. La FGH tipo 1 es una forma autosómica dominante rara que se debe a una mutación en el gen SOS1 que activa la vía RAS/MAPK14. Se desconoce por qué no se asocia con otros defectos del desarrollo, como ocurre con otro tipo de mutaciones SOS1 relacionadas con el SN.
RAS y cáncerEn torno a un 30% de los tumores en humanos tienen mutaciones activadoras de la vía RAS/MAPK6; la prevalencia varía en función del tumor, siendo del 90% en el adenocarcinoma de páncreas, 50% en el colon y tiroides, 30% en el pulmón y 25% en el caso del melanoma. Las mutaciones se detectan con mayor frecuencia en KRAS (en torno a un 85% del total), NRAS (aproximadamente un 15%) y mucho más raramente en HRAS (menos del 1% del total). Estos tres miembros de la familia RAS comparten el 85% de la secuencia de aminoácidos y se expresan en muchos tejidos del organismo, especialmente en el caso de KRAS, detectable en prácticamente la totalidad de las líneas celulares103. Los puntos «calientes» mutacionales de las proteínas RAS producen cambios en los aminoácidos G12, G13 y Q61 que impiden tanto la hidrólisis intrínseca de la forma activa de RAS unida a GTP, como la respuesta a los GAPs. Ello conduce a la activación intrínseca e ilimitada de la cascada metabólica MAPK, entre cuyos efectores se encuentra la proteína RAF, un conocido proto-oncogén. Existen tres isoformas de RAF, de las cuales el BRAF, la más efectiva de ellas dentro de la vía MAPK, está involucrado en el 7% de todos los cánceres humanos y con particular frecuencia en el melanoma, donde se detecta hasta en el 70% de los casos104. La mayoría de las mutaciones se encuentran dentro del dominio cinasa, y la más frecuente de ellas produce una sustitución V600E que activa la vía RAS/MAPK105. Finalmente, se han encontrado mutaciones somáticas en PTPN11 en pacientes con LMMJ, leucemia linfoblástica aguda y leucemia mieloide aguda, por lo que el PTPN11 se considera un oncogén106.
Perspectivas de futuro en DermatologíaLa vía RAS/MAPK está perfectamente definida, por lo que sus alteraciones repercuten en el desarrollo del cáncer y/o en la aparición de síndromes genéticos complejos como los que hemos estudiado. Hace 30 años que se descubrió su implicación en la biología tumoral, y desde entonces se están buscando fármacos antitumorales que la inhiban. Lamentablemente, los esfuerzos encaminados a crear un fármaco inhibidor de la vía RAS/MAPK no han tenido demasiado éxito hasta el momento. En la esfera dermatológica el objetivo principal de los estudios es el melanoma, tumor en el que se detectan mutaciones BRAF y NRAS en el 50-70% y 15-30% de los casos respectivamente107,108, y para el que apenas existen opciones terapéuticas. Parece que las dianas óptimas de la cascada serían las proteínas BRAF y MEK, pero es necesario bloquear también otras rutas metabólicas implicadas en el desarrollo, crecimiento y diseminación del melanoma, como es el caso de las rutas AKT3 y PI3K109. Así, sorafenib, un fármaco inhibidor de RAF aprobado para el tratamiento del hepatocarcinoma avanzado, no se ha mostrado eficaz como agente único en el tratamiento de los melanomas que expresan la mutación BRAF V600E110, probablemente debido a cortocircuitos de retroalimentación que perpetúan la hiperactivación de la vía. Los fármacos inhibidores de MEK y ERK parecen más prometedores, pero de momento no poseen un perfil de biodisponibilidad y toxicidad que permita su empleo109.
En cuanto al tratamiento de las rasopatías se están realizando ensayos clínicos para intentar detener el desarrollo de tumores y mejorar el desarrollo cognitivo de los pacientes con NF1. Hay estudios en curso con sirolimus (un inhibidor de la vía mTOR, que está disregulada en la NF1, la NF2 y la esclerosis tuberosa), imatinib, cediranib (un inhibidor de VEGFR, c-Kit y otras serín proteasas), sorafenib, bevacizumab combinado con RAD001 y lovastatina111. De momento no está claro si los fármacos podrán compensar los mecanismos de retroalimentación de las proteínas inhibidas, ni si los efectos secundarios a largo plazo compensarán el hipotético beneficio de su uso. Sin duda, la escasa prevalencia de las rasopatías dificulta la inclusión de pacientes y disminuye el interés económico de su investigación, de modo que a estos pacientes les queda un largo camino por recorrer antes de que puedan beneficiarse de un tratamiento farmacológico.
Conflicto de interesesLos autores declaran que no tener ningún conflicto de intereses.



