








El síndrome de Kindler es un subtipo de epidermólisis bullosa hereditaria muy rara, causada por la mutación del gen FERMT1 que codifica la proteína kindlina-1.
Clínicamente, se caracteriza por la formación de ampollas inducidas por traumatismo, atrofia cutánea difusa, poiquilodermia, seudosindactilia y fotosensibilidad. En las mucosas, las manifestaciones más frecuentes incluyen conjuntivitis, ectropión, gingivitis hemorrágicas, enfermedad periodontal, pérdida prematura de dientes y colitis severa.
Presentamos los 4 primeros casos con síndrome de Kindler, diagnosticados en el Instituto Nacional de Salud del Niño, Lima, Perú, con el fin de dar a conocer su particular forma de presentación y variedad de manifestaciones clínicas, enfatizando en que este hecho obligó a realizar un manejo multidisciplinario, que permitió un control adecuado de los síntomas y una notable mejoría en su calidad de vida.
Kindler syndrome is a very rare form of bullous epidermolysis. It is a hereditary condition caused by a mutation in the FERMT1 gene that encodes the protein kindlin-1. It is clinically characterized by trauma-induced blistering, diffuse skin atrophy, poikiloderma, pseudosyndactyly, and photosensitivity. The most common mucosal manifestations are conjunctivitis, ectropion, hemorrhagic gingivitis, periodontal disease, premature tooth loss, and severe colitis.
We present the first 4 cases of Kindler syndrome diagnosed at the Instituto Nacional de Salud del Niño in Lima, Peru. These cases highlight the unique clinical presentation and multiple manifestations of this disease and show how a multidisciplinary management approach kept symptoms under control and significantly improved patient quality of life.
El síndrome de Kindler (SK) es una genodermatosis autosómica recesiva descrita por primera vez en 1954 por Kindler1.
Representa un subtipo de epidermólisis bullosa hereditaria (EB) poco frecuente2, se han reportado solo 250 casos a la fecha, a nivel mundial3 y en el Perú, representa el 4,3% de las EB según un reporte en cuya estadística se incluyen los 4 casos presentados en este artículo4.
El SK es causado por la mutación en el gen FERMT1 que codifica la proteína kindlina-1, presente en la piel, el tejido periodontal, el intestino delgado, el colon y el recto5,6; forma parte del citoesqueleto de los queratinocitos basales, interviniendo en la migración, la adhesión y el crecimiento celular5,7; la pérdida de su función resulta en fragilidad cutánea con defectos en la unión de la actina a la matriz extracelular8.
Las manifestaciones clínicas suelen presentarse desde el nacimiento con formación de ampollas inducidas por traumatismo o exposición solar, atrofia cutánea difusa y poiquilodermia de presentación temprana, en áreas fotoexpuestas. La fotosensibilidad mejora con la edad, pero hay una predisposición a desarrollar tumores malignos cutáneos en la edad adulta1,5. El compromiso de mucosas usualmente comienzan en la adolescencia, siendo la mucosa oral la más afectada5,9 con gingivitis hemorrágica, enfermedad periodontal y pérdida prematura de dientes; así mismo pueden presentar conjuntivitis, ectropión, estenosis esofágicas, uretral y anal además de estreñimiento y colitis severa3.
Presentamos el caso de 4 pacientes diagnosticados de SK en los que realizamos un manejo multidisciplinario, logrando un control adecuado de los síntomas y sus complicaciones.
Caso 1Paciente varón, de 13 años de edad, de padres consanguíneos; acude a la consulta por presentar fragilidad en piel y mucosas desde los 5 años de edad que inician como pequeñas erosiones en manos y pies tras mínimos traumatismos, dolor y sangrado de encías, diarrea y estreñimiento intermitente.
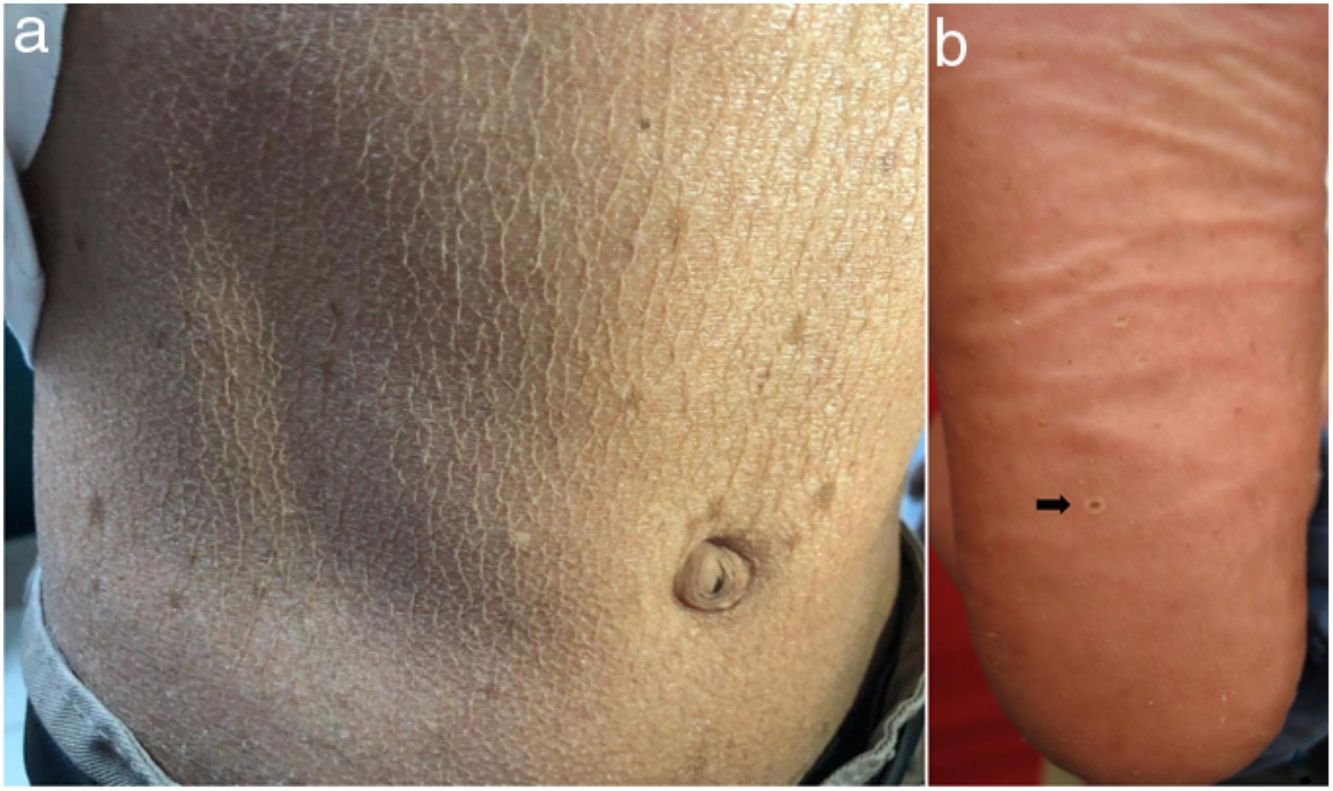
En el examen físico se evidencia piel seca, con áreas atróficas como «papel de cigarillo», hiperpigmención «tipo bronceado» y telangiectasias a predominio de zonas fotoexpuestas; seudosindactilia en manos y pápulas queratósicas puntiformes en plantas (figs. 1 y 2).
 y múltiples efélides a predominio de zonas fotoexpuestas. Se aprecia seudosindactilia en manos de ambos pacientes.")
 Piel seca, con áreas atróficas como «papel de cigarrillo» y múltiples efélides (caso 2). b) Pitting plantar (caso 3).")
En el examen oftalmológico: limitación bilateral en la apertura ocular, ambliopía y estrabismo. En el examen odontológico: disminución a la apertura bucal, labios resecos, con áreas hiperpigmentadas, queilitis angular, estomatitis generalizada y mala higiene oral, encía con hiperplasia gingival (bolsas y seudobolsas periodontales), placa bacteriana, cálculo dental y movilidad grado i en dientes anteriores y caries dental. En el paladar se observan lesiones blanquecinas de aspecto liquenoide (fig. 3).
 Gingivitis, periodontitis e hiperplasia gingival (bolsas y seudobolsas periodontales) en presencia de placa bacteriana y cálculo dental; movilidad grado 1 en dientes anteriores. b) Vista en oclusión postratamiento: se evidencia mejoría clínica significativa. c) Radiografía panorámica evidenciando resorción horizontal de crestas interdentarias.")
Evaluación odontológica del caso 1. a) Gingivitis, periodontitis e hiperplasia gingival (bolsas y seudobolsas periodontales) en presencia de placa bacteriana y cálculo dental; movilidad grado 1 en dientes anteriores. b) Vista en oclusión postratamiento: se evidencia mejoría clínica significativa. c) Radiografía panorámica evidenciando resorción horizontal de crestas interdentarias.
El estudio de inmunofluorescencia (IF) revelo patrón reticular positivo para colágeno iv, laminina 332 y colágeno vii; antikindlina-1 positiva (fig. 4).
, colágeno vii (b) y laminina 332 (c); patrón característico de síndrome de Kindler.")
La microscopia electrónica (ME) mostró segmentos de epidermis con queratinización normal y zonas de clivaje a diferentes niveles de la unión dermoepidérmica, con desmosomas y fibrillas de anclaje presentes (fig. 5).
. Cortes ultrafinos seriados de 100nm de espesor mostrando ampollas (flechas) a diferentes niveles de la unión dermo-epidérmica, con desmosomas y fibrillas de anclaje sin alteraciones.")
En cuanto al tratamiento, se recomendó el uso de bloqueadores solares e hidratantes para la piel en forma permanente. Odontología realizó un tratamiento preventivo, paliativo, sintomático y recuperativo con destartraje y curetaje manual-ultrasónico asociado a un cambio en la dieta y suplemento nutricional enfocado al manejo de la desnutrición crónica.
Luego de 3 meses se observó mejoría en el estado de la piel y la salud periodontal, con cambios significativos en el periodontograma.
Caso 2Paciente varón de 11 años de edad, hermano del primer caso, con lesiones cutáneas desde el nacimiento; desde hace 2 años presenta episodios recurrentes de diarrea alternada con estreñimiento y encopresis, lo que le obliga a usar pañal. En el examen físico: piel seca, con áreas atróficas, hiperpigmención y poiquilodermia en rostro, cuello y tórax; seudosindactilia en manos y pies (fig. 1).
En el examen odontológico: labios resecos, discromía, queilitis angular y microstomía. Maxilar superior con hiperplasia gingival, placa bacteriana y cálculo dental; en el maxilar inferior, gran pérdida de soporte óseo a nivel de incisivos centrales y laterales inferiores, con movilidad grado 3. Así mismo, desnutrición crónica y talla baja.
La IF mostró un patrón reticular positivo para colágeno iv, vii y laminina 5, con antikindlina-1 positiva. La ME mostró ampollas a varios niveles en la unión dermoepidérmica. Rx baritada de colon: dolicomegacolon.
Se recomendó el uso de bloqueadores solares e hidratantes en forma permanente, destartraje y curetaje por Odontología. Gastroenterología planteó un tratamiento con laxantes osmóticos y cambios en la dieta para el manejo de la colitis y encopresis, así como suplemento nutricional.
Caso 3Paciente varón de 15 años de edad, nacido de padres no consanguíneos, que vienen en un poblado pequeño de la Amazonía peruana; acude por lesiones ampollares al mínimo roce desde los 2 meses de edad. En el examen físico se evidenciaron discromía y múltiples lentigos en zonas fotoexpuestas y mucosa labial, además de poiquilodermia en la cara, el cuello y el dorso de manos y pies; seudosindactilia y pitting plantar (figs. 2b y 6). Mucosa oral con hipertrofia gingival y caries dental.
 y efélides de predominio en zonas fotoexpuestas, además de conjuntivitis y leve ectropión bilateral de párpado inferior en la imagen del lado derecho.")
Manifestaciones clínicas de los casos 3 y 4. En ambos casos, se evidencia atrofia cutánea, áreas hipo e hipercrómicas con múltiples telangiectasias (poiquilodermia) y efélides de predominio en zonas fotoexpuestas, además de conjuntivitis y leve ectropión bilateral de párpado inferior en la imagen del lado derecho.
La IF mostró colágeno iv, laminina 332 y colágeno vii en patrón reticular y la ME, ampollas por citólisis en la capa basal, la lámina lúcida y la lámina densa con filamentos y fibrillas de anclaje presentes.
El tratamiento incluyó el uso de bloqueadores solares e hidratantes. Destartraje y curetaje, además de evaluación por nutrición y psicología.
Caso 4Niña de 10 años de edad, hermana del caso 3, quien desde los 6 meses presenta lesiones ampollares que dejan cicatrices discrómicas, con atrofia cutánea, poiquilodermia y efélides de predominio en zonas fotoexpuestas; seudosindactilia en manos, hipertrofia gingival y caries dental. En el examen oftalmológico: conjuntivitis y leve ectropión bilateral de párpado inferior (fig. 6).
La IF y la ME mostraron hallazgos compatibles con SK. El abordaje del paciente incluyó el uso permanente de lágrimas artificiales y lentes con protección UVB-UVA.
DiscusiónEl SK es una entidad autosómica recesiva5; la mayoría de los pacientes reportados son descendientes de padres consanguíneos (casos 1 y 2); pero también puede presentarse como mutación espontánea10. En los hermanos de los casos 3 y 4, los padres niegan parentesco; sin embargo, proceden de pueblos pequeños y fronterizos; si asumimos que en los 4 casos los padres son portadores heterocigotos asintomáticos, la probabilidad de tener un hijo con SK en cada embarazo es del 25%3, de allí la importancia del consejo genético a padres y pacientes.
En el SK la mutación del gen FERMT1 origina la pérdida de su función, conduciendo al despegamiento entre queratinocitos, duplicación de la lámina densa y zonas de clivaje a diferentes niveles en la unión dermoepidérmica, que se expresa desde el nacimiento5,7. Los 4pacientes presentaron fragilidad cutánea desde los primeros años de vida, la cual fue disminuyendo, dejando atrofia cutánea y poiquilodermia de predominio en zonas fotoexpuestas, además de seudosindactilia no tributaria de tratamiento quirúrgico al no comprometer la funcionalidad3. El pitting plantar se evidenció en todos nuestros pacientes.
La formación de ampollas y la fotosensibilidad mejoran con la edad, mientras que la poiquilodermia y la atrofia cutánea continúan evolucionando3. Alrededor del 10% de los pacientes con SK pueden desarrollar tumores malignos cutáneos1,11, como el carcinoma escamocelular después de los 45 años de edad5. El mecanismo de carcinogénesis podría estar relacionado con los niveles bajos de factor de crecimiento transformante-β y la señalización de Wnt aumentada, hallazgos encontrados en humanos y ratones con SK12,13. El uso de protector solar y métodos de barrera en estos pacientes es obligatorio para minimizar el riesgo de carcinogénesis, sobre todo en países con alto nivel de radiación solar, como el nuestro3,13.
El compromiso de mucosas es frecuente, siendo la mucosa oral la más afectada (85%) con periodontitis, hiperplasia gingival, pérdida prematura de los dientes, caries y halitosis14, generándose un círculo vicioso entre mala higiene y enfermedad periodontal. Los 4 pacientes presentaron alteraciones en la mucosa oral, recibiendo un tratamiento preventivo-paliativo y recuperativo dirigido a la afección crónica del tejido periodontal, con una evolución favorable pero de carácter inestable y transitoria, por lo que es necesario continuar con terapia periodontal de soporte, incidiendo en la higiene oral.
La afección de mucosa ocular, esofágica, anal y urogenital es más frecuente después de los 10 años5. El caso 4 presentó conjuntivitis y ectropión leve en el párpado inferior, el cual fue manejado por Oftalmología. El compromiso intestinal en forma de colitis por epiteliosis (15%)5 es más severo en ausencia completa de kindlina-1; la ausencia parcial puede ser compensada por isoformas de kindlina-1 intestinal, por lo que no todos los pacientes tienen síntomas digestivos6. En los 2primeros casos se reportaron episodios de colitis, que en el caso 2 estuvo asociada a encopresis, lo que condicionó un cuadro de desnutrición crónica y talla baja.
El diagnóstico del SK es clínico, apoyado en la IF que muestra un patrón de tinción intenso, amplio y reticulado en la membrana dermoepidérmica con anticuerpos contra laminina 332 y colágeno tipo iv-vii3,15,16; la inmunotinción positiva para antikindlina-1 también se considera diagnóstica3. En la ME se evidencian zonas de clivaje a diferentes niveles: intraepidérmico, intralámina lúcida y sublámina densa3,17, hallazgo característico de esta patología18. Todos los hallazgos anteriormente descritos fueron evidenciados en nuestros 4 pacientes.
Finalmente, el estudio genético molecular permite confirmar el diagnóstico, identificando la mutación del gen FERMT13,5; no fue viable realizar este examen en nuestros pacientes, lo que constituye una de las limitantes del estudio.
ConclusiónEl SK es un subtipo de EB muy pocas veces descrito en la literatura médica. Presentamos los 4 primeros casos reportados en nuestro país, con el fin de dar a conocer su particular forma de presentación y la variedad de manifestaciones clínicas encontradas; enfatizando en que este hecho obliga a que estos pacientes sean manejados por un equipo multidisciplinario, con el objetivo de lograr un diagnóstico oportuno, un control adecuado de sus manifestaciones y la prevención de complicaciones; mejorando así el pronóstico y la calidad de vida de nuestros pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los Dres. Roxana Lipa Chancolla, Luis Claudio Huamaní Huayhua, Gilmer Torres Ramos y Rosario Loaiza de la Cruz.



