









Las mastocitosis constituyen un grupo heterogéneo de enfermedades caracterizadas por la proliferación clonal de mastocitos en distintos órganos, siendo la localización cutánea la más frecuente. Es «una enfermedad rara o poco frecuente», y afecta a todos los grupos de edad, si bien suele aparecer en la primera década de la vida o entre la segunda y la quinta década de la vida, con una distribución similar por sexos. En los últimos años se han realizado grandes avances en el conocimiento fisiopatogénico del trastorno: las mutaciones somáticas del gen c-kit y la presencia de alteraciones inmunofenotípicas en los mastocitos son elementos importantes en la fisiopatogenia de las mastocitosis. Las manifestaciones clínicas son variadas y las lesiones cutáneas son la clave diagnóstica en la mayoría de los pacientes.
Mastocytosis is a term used to describe a heterogeneous group of disorders characterized by clonal proliferation of mast cells in various organs. The organ most often affected is the skin. Mastocytosis is a relatively rare disorder that affects both sexes equally. It can occur at any age, although it tends to appear in the first decade of life, or later, between the second and fifth decades. Our understanding of the pathophysiology of mastocytosis has improved greatly in recent years, with the discovery that somatic c-kit mutations and aberrant immunophenotypic features have an important role. The clinical manifestations of mastocytosis are diverse, and skin lesions are the key to diagnosis in most patients.
La mastocitosis es «una enfermedad rara o poco frecuente», de acuerdo con la Comisión Europea de Salud Pública, enfermedades crónicas debilitantes o potencialmente mortales con una prevalencia inferior a 5 casos por 10.000 habitantes; en el caso de las mastocitosis se estima una prevalencia de 9 casos por 100.0001. En 2 estudios epidemiológicos recientes se publican unas prevalencias puntuales de 9,2 por 100.000 y 13 por 100.000 de mastocitosis sistémica indolente en mayores de 15 años2,3; en el Hospital General de Albacete la prevalencia de mastocitosis sistémica indolente (MSI) en adultos ofrece resultados similares con 11,6 casos por 100.000 habitantes (datos no publicados), y el Instituto de Estudios de Mastocitosis de Castilla-La Mancha (CLMast) sugiere que la prevalencia de las formas con afectación cutánea se encuentra en torno a 0,2 casos por cada 100.000 habitantes y año4. Sin embargo, estos datos son estimativos y hay que tomarlos con cautela, ya que se desconoce la prevalencia exacta de formas como los mastocitomas solitarios o de pacientes con mastocitosis sistémica sin lesión cutánea, en los que la enfermedad cursa con anafilaxia.
Las mastocitosis afectan a todos los grupos de edad, si bien la enfermedad suele aparecer en la primera década de la vida, en más del 50% de los casos en los 2 primeros años, siendo el inicio congénito menos frecuente5, o entre la segunda y quinta década de la vida, con una distribución similar por sexos5,6.
Se han descrito casos familiares, con al menos un familiar de primer grado afecto (2-4%)7,8, la mayoría relacionados con diversas mutaciones germinales del gen c-kit9,10.
Las mastocitosis constituyen un grupo heterogéneo de enfermedades con un rasgo común: la proliferación y acumulación de mastocitos (MC) patológicos en distintos tejidos, afectando con frecuencia la piel, la médula ósea y el tracto gastrointestinal, así como la presencia de síntomas secundarios a la acción de los mediadores liberados tras la activación mastocitaria en la mayoría de pacientes11–14. Son enfermedades del sistema hematopoyético de carácter clonal, lo que se ha establecido por la demostración de mutaciones en el receptor mastocitario de membrana KIT en la mayoría de los adultos afectados15, y también en una proporción elevada de casos pediátricos9.
Existen diferentes formas de mastocitosis atendiendo a la edad de aparición (mastocitosis pediátricas y del adulto), el número de órganos afectos (mastocitosis cutáneas y sistémicas) y el comportamiento clínico (mastocitosis indolentes y agresivas). De acuerdo con el momento de inicio de la enfermedad, las mastocitosis se dividen en pediátricas y del adulto, y suelen presentar un comportamiento diferente, de modo que en un porcentaje elevado de casos los pacientes pediátricos solo presentan lesiones cutáneas, sin apenas asociar síntomas secundarios a la liberación de mediadores mastocitarios y las lesiones tienden a desaparecer alrededor de la pubertad16–18: prácticamente el 100% de los mastocitomas solitarios lo hacen, mientras que las formas clínicas, con lesiones más extensas, pueden persistir en un 30-50% de casos8. Por el contrario, los pacientes que desarrollan la enfermedad en la edad adulta presentan en su mayoría afectación sistémica (demostrada por la presencia de mastocitos patológicos en la médula ósea o cualquier otra localización extracutánea)13,14,19,20, que persiste a lo largo de la vida. No obstante, esta clasificación puede resultar artificiosa, pues todos los MC se originan en la médula ósea y, por tanto, conceptualmente la mastocitosis es siempre una enfermedad sistémica.
Fisiopatología de las mastocitosisEl MC es una célula hematopoyética derivada de la célula progenitora mieloide pluripotencial21. Los precursores mastocitarios emigran desde la médula ósea a la sangre y de aquí a los tejidos, donde terminan su diferenciación, adquiriendo las características morfológicas, inmunofenotípicas y funcionales del tejido donde se localizan, al mismo tiempo que mantienen su capacidad proliferativa11,21.
El protooncogén c-Kit, localizado en el cromosoma 4q12 en humanos22, codifica una glucoproteína de superficie que actúa como receptor transmembrana con actividad tirosina cinasa intrínseca, la proteína KIT (adscrita al grupo de diferenciación CD117), que se expresa en los precursores hematopoyéticos CD34+de médula ósea, sangre periférica y sangre del cordón umbilical. La expresión del KIT se pierde durante el proceso de maduración en la mayoría de células hematopoyéticas, pero no en los MC, células en las que ejerce un papel fundamental en su proliferación, supervivencia y función23. La expresión del KIT no se restringe a las células hematopoyéticas, sino que también lo expresan los melanocitos o las células intersticiales de Cajal del tracto gastrointestinal, entre otras4.
La estructura del receptor KIT se organiza en 5 dominios: uno extracelular glucosilado, el transmembrana, el yuxtamembrana y 2 citoplasmáticos tirosina cinasa (fig. 1)23,24. Los precursores mastocitarios maduran por la activación del receptor KIT, cuando el dominio extracelular del KIT se une a su ligando, el Stem cell factor (SCF), sintetizado fundamentalmente por las células del estroma. Esta interacción del KIT con su ligando es fundamental en el desarrollo y maduración mastocitaria25, estimulando también la adhesión, la migración, la supervivencia y la liberación de mediadores por los MC maduros24.
.")
Estructura del receptor KIT23,24. En rojo, localización de alguna de las mutaciones activantes, en el dominio yuxtamembrana y en el tirosina cinasa ii (D816V, mutación más frecuente en mastocitosis).
Los MC se encuentran en prácticamente todos los órganos y tejidos, pero son especialmente numerosos en la piel, el sistema respiratorio, en el tracto digestivo y genitourinario, sobre todo en la proximidad de los vasos sanguíneos, linfáticos y alrededor de los nervios periféricos. Su localización en las superficies próximas al medio externo se debe a que son células efectoras del sistema inmunológico, tanto de la inmunidad adquirida como innata26, activándose a través de los receptores de alta afinidad para la IgE (Fc¿RI) que expresan en superficie, aunque también pueden hacerlo por otros mecanismos tanto inmunológicos (como los receptores de IgG [FcγR]27, receptores para moléculas del sistema del complemento, como el C3aR y el C5aR [CD88])28, el receptor de alta afinidad para el factor de crecimiento nervioso [TrkA]28,29 o receptores de tipo toll-like y nucleotide-binding oligomerization domain30) como no inmunológicos (fármacos y factores físicos31).
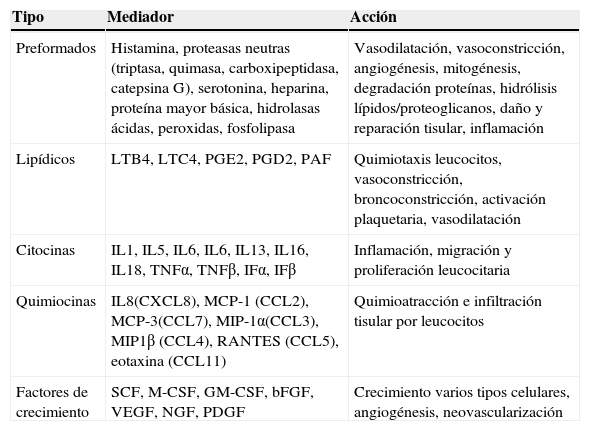
Además intervienen en otras funciones como son la presentación de antígenos, la angiogénesis, la cicatrización de las heridas, la remodelación tisular, la fibrosis, el rechazo de injertos y la vigilancia tumoral32. Su actividad la ejercen por medio de la liberación de múltiples mediadores, algunos preformados y almacenados en gránulos y otros sintetizados y liberados tras el estímulo inductor (tabla 1).
Mediadores mastocitarios y efectos fisiológicos
| Tipo | Mediador | Acción |
|---|---|---|
| Preformados | Histamina, proteasas neutras (triptasa, quimasa, carboxipeptidasa, catepsina G), serotonina, heparina, proteína mayor básica, hidrolasas ácidas, peroxidas, fosfolipasa | Vasodilatación, vasoconstricción, angiogénesis, mitogénesis, degradación proteínas, hidrólisis lípidos/proteoglicanos, daño y reparación tisular, inflamación |
| Lipídicos | LTB4, LTC4, PGE2, PGD2, PAF | Quimiotaxis leucocitos, vasoconstricción, broncoconstricción, activación plaquetaria, vasodilatación |
| Citocinas | IL1, IL5, IL6, IL6, IL13, IL16, IL18, TNFα, TNFβ, IFα, IFβ | Inflamación, migración y proliferación leucocitaria |
| Quimiocinas | IL8(CXCL8), MCP-1 (CCL2), MCP-3(CCL7), MIP-1α(CCL3), MIP1β (CCL4), RANTES (CCL5), eotaxina (CCL11) | Quimioatracción e infiltración tisular por leucocitos |
| Factores de crecimiento | SCF, M-CSF, GM-CSF, bFGF, VEGF, NGF, PDGF | Crecimiento varios tipos celulares, angiogénesis, neovascularización |
bFGF: factor de crecimiento fibroblástico básico; CCL: quimiocina (C-C motif) ligando; GM-CSF: factor estimulante de las colonias de macrófagos y granulocitos; IF: interferón; IL: interleucina; LTB4: leucotrieno B4; LTC4: leucotrieno C4; MCP: proteina quimioatractora de monocitos; M-CSF: factor esimulante de las colonias de macrófagos; MIP: proteina inflamatoria macrofágica; NGF: factor de crecimiento nervioso; PAF: factor activador plaquetario; PDGF: factor de crecimiento derivado de las plaquetas; PG: prostaglandina; RANTES: citocina expresada y secretada por el linfocito T normal en función de su grado de activación; SCF: factor de células madre; TNF: factor de necrosis tumoral; VEGF: factor de crecimiento del endotelio vascular.
Fuente: Akira et al.11.
En los últimos años se han producido grandes avances en los conocimientos fisiopatogénicos, que han permitido desarrollar nuevas técnicas diagnósticas y estrategias terapéuticas, y establecer nuevas clasificaciones. Además, se han producido cambios legislativos nacionales y europeos, que han permitido incluir esta entidad dentro de las enfermedades raras o poco frecuentes y el desarrollo de unidades monográficas y centros de referencia que trabajan en red (Red Española de Mastocitosis [REMA]; integrada dentro de la red europea (European Competence Network on Mastocytosis [ECNM])12 y que establecen documentos de consenso en diagnóstico y tratamiento para asegurar el derecho a la salud de estos pacientes20.
Las mutaciones somáticas del gen c-kit y la presencia de alteraciones inmunofenotípicas en los MC resultan claves en la fisiopatogenia.
Mutaciones de c-KitSe han descrito múltiples mutaciones en el protooncogén c-Kit capaces de activar el receptor de forma independiente a su ligando, lo que se conoce como mutaciones activantes33,34. La presencia de estas mutaciones se ha relacionado con la fisiopatología de tumores estromales gastrointestinales, el seminoma, el melanoma y por supuesto linfomas, procesos mieloproliferativos y las mastocitosis9,33–35.
La mayoría de las mutaciones en la mastocitosis se sitúan en 2 regiones del c-kit, en el exón 11 que codifica el dominio yuxtamembrana y, sobre todo, en el exón 17 que codifica el dominio tirosina cinasa 29. Estas mutaciones inducen una activación constitutiva independiente del ligando, lo que determina una proliferación clonal. En esa localización las mutaciones más frecuentes son puntuales de sentido erróneo (missense) en el exón 17 (codones 816 y 815, la más común la sustitución de valina por aspártico en el dominio catalítico del KIT, Asp 816 Val o D816V)11. Estas mutaciones se detectan en más del 90% de los casos de las mastocitosis desarrolladas en adultos15,33,34; sin embargo, en las formas pediátricas los estudios ofrecen resultados variables (0-83%), si bien se trata de series cortas de pacientes y focalizados en la detección de mutaciones en el codón 81633,36–39.
Sotlar et al. fueron los primeros en estudiar sistemáticamente la mutación en el codón 816 en niños con mastocitosis pediátrica, encontrando que alrededor del 40% de los pacientes la presentaban37. Más recientemente, Bodemer et al.9 realizaron un estudio en 50 niños (0-16 años) con mastocitosis mediante secuenciación del c-Kit en muestras cutáneas, detectando mutaciones activantes de c-Kit en el 86% de los pacientes, en el codón 816 en el 42% de los casos y en otras localizaciones distintas del exón 17 (dominio extracelular y yuxtamembrana) en el 44%, sin poder establecer una correlación entre el tipo de mutación y el fenotipo, excepto la ausencia de mutaciones en el codón 816 cuando la edad de inicio era entre 3 y 16 años. Todos estos estudios apoyan el carácter clonal de las mastocitosis pediátricas, a pesar de su tendencia a la regresión espontánea en muchos casos9. Por otra parte, es probable que las diferentes mutaciones halladas se puedan correlacionar con ciertos tipos de la enfermedad y tener implicaciones terapéuticas, aunque a día de hoy aún no se pueda confirmar esta hipótesis.
Por lo tanto, la presencia de mutaciones activantes en el gen c-Kit sería un requisito necesario para el desarrollo de las mastocitosis, y la diversidad fenotípica podría estar relacionada con la combinación con otras mutaciones adquiridas u otros polimorfismos genéticos heredados39.
Inmunofenotipo de los mastocitos en las mastocitosisEl uso de la citometría de flujo, técnica que es capaz de identificar y cuantificar células presentes en muy baja frecuencia en una muestra como ocurre con los MC, ha permitido identificar un inmunofenotipo específico de los MC patológicos de médula ósea y otros tejidos en las mastocitosis: la expresión de CD25, receptor de la cadena α de la interleucina 240. Su presencia constituye un marcador casi patognomónico de las mastocitosis sistémicas (excepto en las mastocitosis sistémicas bien diferenciadas)41, y se relaciona con la activación y proliferación celular42.
Nunca se detecta en MC de sujetos sanos o de pacientes con otras enfermedades (hematológicas o no), siendo la única excepción los síndromes hipereosinofílicos FIP1L1/PDGFR (alfa o beta) con MC clonales43. Por otra parte, recientemente se ha evaluado la utilidad del CD30 como marcador expresado en la mayoría de mastocitosis sistémicas agresivas44 y de las mastocitosis sistémicas bien diferenciadas45.
ClínicaLas manifestaciones clínicas se relacionan con la liberación masiva o crónica de los mediadores mastocitarios (en la mayoría de los casos, formas pediátricas y no agresivas del adulto)46, la infiltración tisular o con la presencia de otro trastorno hematológico asociado. Entre estos síntomas secundarios a la liberación de mediadores mastocitarios se encuentran el prurito, el enrojecimiento, acompañado o no de palpitaciones y/o cefalea, la formación de ampollas sobre las lesiones cutáneas en algunas formas pediátricas (fig. 2), sobre todo en los primeros años de vida, el dolor abdominal, la diarrea, la hipotensión, la anafilaxia y los síntomas neuropsiquiátricos (irritabilidad, falta de atención)46. En las formas pediátricas los síntomas suelen ser más intensos en los 18 meses siguientes a la aparición de las lesiones cutáneas4.

Se ha descrito un porcentaje de anafilaxia del 6-9% en las formas pediátricas y del 20-49% de las mastocitosis del adulto47–49, cifras ambas por encima de la frecuencia de anafilaxia descrita en población general50,51, aunque con una prevalencia de reacciones alérgicas mediadas por IgE similares48,49.
La anafilaxia es una frecuente forma de presentación de las mastocitosis sistémicas del adulto sin lesión cutánea, predominando la sintomatología cardiovascular tras la picadura de himenóptero en pacientes varones31,52,53.
La aparición aguda de síntomas generalmente es producida por diversos desencadenantes. Los más característicos son los agentes físicos, como la fricción de las lesiones o el calor, el estrés, los fármacos y el veneno de himenópteros, entre otros (tabla 2)31.
Factores desencadenantes para la liberación de mediadores mastocitarios en las mastocitosis
| Agentes físicos |
| Calor, cambios de temperatura |
| Fricción de los mastocitomas |
| EndoscopiasManipulación de tracto gastrointestinal (p. ej. durante cirugía) |
| Factores emocionales |
| Estrés, ansiedad |
| Fármacos |
| Antiinflamatorios no esteroideos |
| Opioides |
| Anestésicos |
| Contrastes radiológicos iodados intravenososInterferón α2b |
| Picaduras |
| Himenópteros |
La acción de los mediadores mastocitarios liberados (histamina, heparina, triptasa y sobre todo citoquinas como el factor de necrosis tumoral α, la interleucina 1 y la interleucina 6)54 pueden producir alteraciones óseas como osteoporosis, detectada en alrededor del 18% de las MSI55 y esclerosis ósea difusa (60% de las mastocitosis sistémicas agresivas) —REMA, datos no publicados— habiéndose detectado una correlación positiva entre niveles elevados de metabolitos de histamina en orina y el riesgo de osteoporosis56. También por acción de estos mediadores los pacientes pueden manifestar síntomas constitucionales, que son casi exclusivos de las formas agresivas de la enfermedad57.
La infiltración tisular, sobre todo en las formas agresivas de la enfermedad, puede dar lugar a signos y síntomas secundarios como: hepatoesplenomegalia, adenopatías, dolor abdominal, o las alteraciones en la circulación portal y ascitis13.
A pesar de que las manifestaciones son heterogéneas, la mayoría de pacientes presentan un curso clínico indolente. La piel es el órgano que se afecta con mayor frecuencia en prácticamente el 100% de las mastocitosis pediátricas y en alrededor del 85% de las del adulto: es decir, la ausencia de lesiones cutáneas no excluye la existencia de una mastocitosis13.
Los signos y síntomas que pueden llevar a un paciente con mastocitosis a requerir atención médica son los siguientes14:
- 1)
Lesiones cutáneas, ya sean percibidas por el paciente o sus familiares (en el caso de los niños), o detectadas en el curso de una exploración por otros motivos.
- 2)
Síntomas secundarios a la acción de la liberación de mediadores mastocitarios, como prurito, dolor abdominal, diarrea, anafilaxia con colapso vascular en ausencia de urticaria/angioedema y desencadenados por picadura de himenóptero58,59; aunque estos síntomas pueden aparecer con o sin desencadenante identificado y mediados o no por IgE.
- 3)
Astenia y pérdida de peso, acompañadas de hepatomegalia o esplenomegalia y alteraciones hematológicas (anemia, leucocitosis, trombocitosis).
- 4)
Fracturas patológicas por osteoporosis avanzadas en pacientes sin otros factores de riesgo de osteoporosis (especialmente varones jóvenes o de edad media).
- 5)
Síntomas gastrointestinales inespecíficos sugestivos de colitis o esplenomegalia.
- 6)
O pacientes diagnosticados de un síndrome mielodisplásico o mieloproliferativo y en los que en el estudio se detecta una mutación del c-Kit14.
Las lesiones cutáneas son la clave diagnóstica en muchos pacientes; un dermatólogo experto diagnostica la mastocitosis cutánea por los hallazgos clínicos con un porcentaje de éxito superior al 90%4. Incluso se han empleado técnicas diagnósticas no invasivas como la dermatoscopia, describiéndose 4 patrones (marrón homogéneo, amarillo homogéneo, reticular vascular y reticular pigmentado), que si bien no son específicos ayudan al diagnóstico60. La presencia de las lesiones cutáneas nos permite establecer el diagnóstico provisional de mastocitosis en la piel que se confirmará mediante biopsia lesional y estudio histopatológico20,61,62 (con tinciones panópticas, metacromáticas y/o inmunohistoquímica empleando anticuerpos frente a triptasa y/o KIT) (fig. 3), aunque en los mastocitomas en muchas ocasiones basta con el diagnóstico clínico. El estudio histopatológico de la piel mostrará un infiltrado mastocitario en la dermis con 4 patrones: el perivascular en la dermis papilar y reticular superior, el patrón en sábana en la dermis superior, el intersticial y el nodular63. El patrón de infiltración mastocitaria cutánea no tiene valor predictivo en cuanto al riesgo de afectación sistémica, y se correlaciona solo de forma parcial con la morfología clínica63.
. B. Gránulos metacromáticos en el citoplasma (azul de toluidinax200).")
El diagnóstico definitivo de mastocitosis cutánea se define por la tríada de lesión cutánea típica, confirmación histopatológica de infiltrados focales mastocitarios en la dermis y ausencia de criterios de afectación sistémica13.
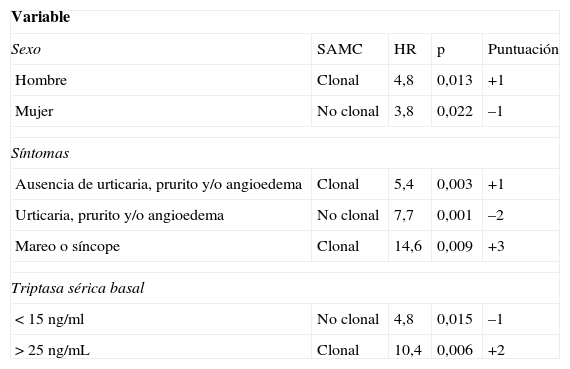
Por otra parte, en los pacientes sin lesión cutánea típica, pero en los que se sospecha una posible mastocitosis debido a que han presentado una anafilaxia, la REMA ha desarrollado una herramienta de despistaje para predecir la clonalidad de los MC (tienen una mutación de KIT [D816V] y/o expresión de CD25+)58, que ha sido aceptada por la ECNM64, basándose en datos clínicos y analíticos del paciente (tabla 3).
Modelo de puntuación para predecir la clonalidad de mastocitos de médula ósea y mastocitosis sistémica en casos con síntomas secundarios a activación mastocitaria
| Variable | ||||
| Sexo | SAMC | HR | p | Puntuación |
| Hombre | Clonal | 4,8 | 0,013 | +1 |
| Mujer | No clonal | 3,8 | 0,022 | –1 |
| Síntomas | ||||
| Ausencia de urticaria, prurito y/o angioedema | Clonal | 5,4 | 0,003 | +1 |
| Urticaria, prurito y/o angioedema | No clonal | 7,7 | 0,001 | –2 |
| Mareo o síncope | Clonal | 14,6 | 0,009 | +3 |
| Triptasa sérica basal | ||||
| <15ng/ml | No clonal | 4,8 | 0,015 | –1 |
| >25ng/mL | Clonal | 10,4 | 0,006 | +2 |
Puntuación<2: baja probabilidad de SAMC-c; puntuación>2: alta probabilidad de SAMC-c.; sensibilidad: 0,92; especificidad: 0,81; VPP: 0,89; VPN: 0,87.
HR: hazard ratio; SAMC: síndrome de activación mastocitaria; SAMC-c: síndrome de activación mastocitaria clonal; VPN: valor predictivo negativo; VPP: valor predictivo positivo.
Fuente: Alvarez-Twose et al.58.
Los criterios diagnósticos de la OMS para establecer el diagnóstico de MS11,13,14,20 han sido evaluados por la REMA por medio de estudios prospectivos. Se han dividido en criterios directos, capaces de detectar una lesión anatómica anormal (los agregados de MC) o de identificar la presencia de MC anormales por su morfología, expresión de moléculas de superficie (CD25) o la presencia de marcadores moleculares anormales (mutaciones del c-Kit) y criterios indirectos, que permiten sospechar la existencia de una MS19 (tabla 4).
Criterios diagnósticos de mastocitosis sistémica
| Criterios directos | Mayor | Agregados multifocales de más de 15 MC en cortes histológicos y/o en extensiones de médula ósea. Este criterio se puede aplicar a otros tejidos |
| Menores | Más del 25% de MC morfológicamente atípicos en las extensiones de médula ósea | |
| Expresión del antígeno CD25 en MC de médula ósea con o sin expresión de CD2 | ||
| Detección de una mutación de c-Kit en el exón 17 u otro diferentea | ||
| Criterios indirectos | Aumento de la triptasa sérica basalb | |
| Presencia de mastocitosis cutáneac |
MC: mastocitos.
Fuente: García-Montero et al.19.
En las mujeres con mutación de c-Kit negativa, demostración de clonalidad mediante la prueba de HUMARA, análisis de la clonalidad de un tejido basado en el estudio del patrón de inactivación del gen del receptor androgénico humano localizado en el cromosoma X, lo que se produce aleatoriamente (policlonal) en los diferentes tejidos y de forma clonal en las neoplasias.
Un estudio de médula ósea que pueda permitir el diagnóstico y pronóstico de una mastocitosis debe incluir: la citología; la histología convencional con tinciones clásicas (hematoxilina-eosina), metacromáticas como el Giemsa o el azul de toluidina y técnicas inmunohistoquímicas de mayor sensibilidad con anticuerpos frente a triptasa (fig. 4) y al receptor KIT65; el estudio del inmunofenotipo de MC mediante citometría de flujo11,13,14,20,46,66; y la detección de mutaciones de c-Kit en MC purificados de médula ósea, así como en otras líneas hematopoyéticas19.
.")
Ya que los MC están unidos al estroma, el diagnóstico citológico requiere que las extensiones de médula ósea contengan un número suficiente de partículas medulares para permitir un adecuado examen morfológico. Los MC en las mastocitosis se caracterizan por presentar una morfología alargada, un citoplasma con una menor densidad granular que los mastocitos normales, una distribución anómala de los gránulos y fusión granular, el núcleo es oval, e incluso en las formas agresivas se pueden observar células binucleadas13.
La lesión habitual en la médula ósea son los infiltrados densos, de más de 15 MC, multifocales y de localización paratrabecular o perivascular65, siendo frecuente en las formas agresivas la presencia de fibrosis.
El porcentaje de MC en la médula ósea es bajo tanto en pacientes con mastocitosis sistémica como en la población general, un 0,27% de media frente a un 0,021% respectivamente65. Por ello la citometría de flujo para la detección de estas células y su inmunofenotipo ha supuesto un avance indudable, ya que permite detectar, cuantificar y cualificar las características patológicas de los MC con una mayor sensibilidad, incluso cuando están presentes en la muestra en cantidad muy escasa66–68.
Además, la purificación mediante la técnica de clasificación de células activadas por fluorescencia (fluorescence-activated cell sorting) de MC y otras líneas hematopoyéticas como los neutrófilos, los monocitos y los linfocitos, con una pureza superior al 97%, permite establecer el patrón de mutaciones de c-Kit (restringida al mastocito o multilineal) que está directamente asociado al pronóstico de la enfermedad15,44,55.
En la práctica habitual de la REMA no se realizan de modo rutinario estudios de médula ósea a las mastocitosis pediátricas, salvo casos excepcionales con síntomas muy graves secundarios a la liberación de mediadores mastocitarios, con niveles elevados de triptasa que asocien hepatoesplenomegalia y/o citopenias.
Triptasa séricaLas triptasas son proteasas localizadas en los gránulos mastocitarios y, en menor cantidad, en los basófilos sanguíneos; se han descrito varias isoformas en humanos, todas ellas codificadas por genes localizados en el cromosoma 16 (entre otras la α-triptasa y la β-triptasa).
La α-triptasa es liberada de forma constitutiva al plasma, mientras que la β-triptasa solo tras la activación de los MC como ocurre en las situaciones asociadas con la degranulación mastocitaria masiva69.
La determinación de los valores de triptasa total en plasma o suero ha supuesto uno de los mayores avances en el diagnóstico y seguimiento de las mastocitosis. Para ello se emplea un inmunoanálisis comercial (ImmunoCAP Tryptase, Thermo Fisher Scientific Inc.) capaz de cuantificar la triptasa total, sin distinguir entre formas maduras o precursores ni entre las isoformas α o β en los fluidos biológicos.
La OMS ha establecido como un criterio diagnóstico menor de mastocitosis sistémica una cifra de triptasa basal superior a 20ng/ml13, sin embargo debe destacarse que según la experiencia de la REMA en las MSI la cifra de triptasa es inferior a 20ng/ml en el 25% de los casos19.
En los adultos los niveles de triptasa sérica total se han relacionado con la carga total de MC presente en el organismo70, así como con el grado de infiltración mastocitaria en la médula ósea de las mastocitosis sistémicas71. Además, la elevación progresiva de los niveles de triptasa en determinaciones seriadas se relaciona con progresión de la enfermedad y un peor pronóstico55. Sin embargo, en las mastocitosis pediátricas esta relación no es tan clara, aunque se ha descrito que los valores de triptasa más elevados se hallan en niños con formas extensas de afectación cutánea; además, estos tienen un riesgo mayor de presentar episodios de síntomas secundarios a la liberación de mediadores mastocitarios de potencial gravedad72.
También se pueden observar elevaciones de la triptasa sérica en otras situaciones patológicas diferentes de las mastocitosis como: episodios anafilácticos, hemopatías mieloides como las leucemias mieloides agudas, síndromes mielodisplásicos y leucemia mieloide crónica, síndromes hipereosinofílicos en los que se detecta el gen de fusión FIP1L1/PDGFRA junto con la presencia de MC anormales CD25 positivos, y enfermedades no hematológicas como la urticaria crónica y la insuficiencia renal avanzada55.
Otras exploraciones complementarias y algoritmos diagnósticosPara el diagnóstico y seguimiento de un paciente con mastocitosis también se indican las siguientes exploraciones complementarias: hemograma, bioquímica completa, coagulación y metabolitos de histamina (ácido metilimidazol acético) en orina73.
Además, en adultos se realizarán estudios de imagen como la ecografía abdominal (algunos autores lo recomiendan de manera sistemática también en las formas pediátricas, salvo los mastocitomas)46 y densitometría ósea. En algunos casos se realizarán una serie ósea, tomografía axial computarizada o resonancia magnética, para valorar la presencia de visceromegalias, adenopatías y/o esclerosis ósea difusa o parcheada.
Las figuras 5 y 6 muestran el algoritmo diagnóstico a seguir en pacientes con sospecha de mastocitosis sistémica con y sin lesiones cutáneas, respectivamente74.

Algoritmo diagnóstico en pacientes con lesiones cutáneas sospechosas de mastocitosis.
Fuente: Alvarez-Twose et al.58.

Las mastocitosis son un grupo de enfermedades «raras o poco frecuentes» de carácter clonal con manifestaciones variadas, pero la mayoría de pacientes presentan un curso clínico indolente. La piel es el órgano que se afecta con mayor frecuencia. Para la valoración de una posible afectación sistémica se realiza un estudio de médula ósea, excepto en los casos pediátricos, en los que no se indica de modo rutinario, salvo casos excepcionales con alto grado de sospecha clínica.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.
Al Dr. Luis Escribano por su liderazgo, compromiso y motivación en el estudio de las mastocitosis.



