






















La piel puede ser «clave» en el diagnóstico precoz de neoplasias sistémicas. En esta segunda parte se abordan una serie de dermatosis agrupadas por sus características morfológicas, muy diversas, que en un contexto apropiado contribuyen a desenmascarar procesos malignos. Se analizan las lesiones erosivas y ampollares, las pápulas y nódulos inflamatorios, la xerosis, ictiosis, dermatitis exfoliativa generalizada, síntomas como el prurito, alteraciones de la distribución del pelo, trastornos de la sudoración, tumores benignos que pueden formar parte de síndromes heredofamiliares en cuya evolución se puede desarrollar cáncer visceral. Por último se describen las lesiones ungueales y bucales en relación con malignidad sistémica.
En definitiva, se destaca la importancia de la piel en el estudio de las neoplasias sistémicas.
The skin can be key to early diagnosis of systemic malignancies. In the second part of this review, we present various skin conditions that can, in certain contexts, reveal the presence of malignancy. The skin conditions are presented in groups based on a diverse range of morphological characteristics. Specifically, the following groups are analyzed: erosive and blistering lesions; inflammatory papules and nodules; xerosis, ichthyosis, and generalized exfoliative dermatitis; symptoms such as pruritus; abnormal hair distribution patterns; sweating disorders; benign tumors that can form part of hereditary syndromes associated with a risk of visceral cancer; and finally, oral and nail abnormalities.
This review highlights the importance of the skin in the study of systemic malignancies.
La piel nunca debe ser ignorada en las enfermedades sistémicas, es quizás el órgano más accesible y su exploración no requiere técnicas agresivas.
En las neoplasias internas la piel puede ofrecer el primer síntoma guía en el 1% de los pacientes. Esto, junto a una historia clínica detallada, supone una importante alerta de la presencia de un tumor maligno.
Las manifestaciones cutáneas de malignidades internas se pueden producir por invasión directa de la piel por el tumor y por diseminación metastásica, pero existen mecanismos indirectos que inducen la aparición de signos y síntomas cutáneos no relacionados con el tumor primitivo. Algunos de ellos cursan de forma aislada, pero otros forman parte de síndromes paraneoplásicos complejos. Ambos son indicativos de la presencia de una neoplasia subyacente1–7.
Se continúa con la revisión de los signos y síntomas cutáneos que pueden conducir al diagnóstico de una malignidad subyacente, formen parte o no de los síndromes paraneoplásicos. Se enumeran una gran variedad de procesos, algunos de ellos definidos genuinamente como «paraneoplásicos clásicos» (se señalan con un asterisco [*]), pero otros corresponden a un grupo amplio de dermatosis frecuentes observadas en el ámbito cotidiano, habitualmente no indicativas de malignidad, pero que en determinadas situaciones y con ciertas peculiaridades nos pueden alertar de la presencia de una neoplasia y requieren un alto grado de «sospecha clínica». Del clínico depende realizar pruebas complementarias para su estudio o mantener un seguimiento, sobre todo en aquellos en que la asociación a malignidad no tiene una frecuencia significativa o en algunas genodermatosis que en su evolución tienen mayor riesgo de desarrollar determinadas neoplasias. Se señalan con 2 asteriscos (**) los procesos ocasionalmente asociados a una neoplasia interna y con 3 (***) los excepcionalmente asociados a neoplasias internas.
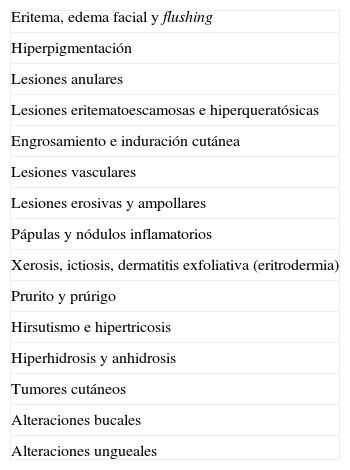
Se optó por agrupar los signos y síntomas según su morfología clínica, enumerándolos de una forma aleatoria (tabla 1). En esta 2.ª parte se exponen las distintas entidades que se enumeran comenzando por el grupo de lesiones erosivas y ampollares.
Signos cutáneos indicadores de posibles neoplasias internas
| Eritema, edema facial y flushing |
| Hiperpigmentación |
| Lesiones anulares |
| Lesiones eritematoescamosas e hiperqueratósicas |
| Engrosamiento e induración cutánea |
| Lesiones vasculares |
| Lesiones erosivas y ampollares |
| Pápulas y nódulos inflamatorios |
| Xerosis, ictiosis, dermatitis exfoliativa (eritrodermia) |
| Prurito y prúrigo |
| Hirsutismo e hipertricosis |
| Hiperhidrosis y anhidrosis |
| Tumores cutáneos |
| Alteraciones bucales |
| Alteraciones ungueales |
La presencia de erosiones y ampollas en la piel y en las mucosas orientan a cuadros como el liquen plano, el eritema exudativo multiforme y las enfermedades ampollares autoinmunes. A veces se imbrican todo este tipo de lesiones en un mismo proceso, como ocurre en el pénfigo paraneoplásico8,9, en que la malignidad subyacente puede diagnosticarse tanto en niños10 como en adultos (tabla 2).
Dermatosis con lesiones erosivas y ampollares y posible asociación a neoplasia interna
| Pénfigo paraneoplásico* |
| Otras enfermedades erosivo-ampollosas: |
| Penfigoide ampolloso*** |
| Penfigoide cicatricial*** |
| Dermatitis herpetiforme*** |
| Dermatitis ampollosa IgA lineal*** |
| Epidermólisis ampollosa adquirida*** |
| Pénfigo foliáceo*** |
| Eritema exudativo multiforme*** |
*Procesos genuinamente paraneoplásicos; ***procesos excepcionalmente asociados a neoplasia interna.
El pénfigo paraneoplásico posee unos rasgos peculiares que lo pueden diferenciar del pénfigo vulgar: la estomatitis es intensa, dolorosa y persistente, se afectan otras mucosas, puede acompañarse de una erupción cutánea polimorfa, vesiculosa y liquenoide que a veces remeda un eritema exudativo multiforme, una necrólisis epidérmica tóxica, una eritrodermia o un liquen plano. Las lesiones acrales con afectación palmoplantar y la paroniquia son frecuentes y poco evidentes en el pénfigo vulgar (figs. 1 y 2).
.")
.")
La biopsia cutánea muestra una dermatitis de interfase, vacuolización de la capa basal, queratinocitos necróticos, hendiduras suprabasales y acantólisis. El componente inflamatorio liquenoide puede ser mayor que en el pénfigo convencional. La inmunofluorescencia directa detecta depósitos de IgG y C3 en los espacios intercelulares de la epidermis y en la unión dermoepidérmica, pero también un rasgo peculiar es que puede ser negativa11. En sangre periférica se identifican anticuerpos antiplaquina circulantes.
En dos tercios de los casos se diagnostica la neoplasia antes del comienzo del pénfigo, asociándose más frecuentemente a linfoma no Hodgkin, a leucemia linfoide crónica y a enfermedad de Castleman, esta última más frecuente en niños. Con menor incidencia se asocian timoma, macroglobulinemia de Waldenstrom, adenocarcinoma de pulmón, linfoma folicular, sarcoma retroperitoneal y ocasionalmente carcinoma hepatocelular12.
En el tercio restante la neoplasia surge después del pénfigo, por lo que se aconseja investigarla en pacientes con lesiones mucosas persistentes refractarias al tratamiento. El fallo respiratorio por bronquiolitis obliterante es un evento común y terminal en estos pacientes, que pueden fallecer entre un mes y 2 años después del diagnóstico13,14.
Otras enfermedades ampollares autoinmunes que se enumeran a continuación son fuente de debate, en cuanto que suponen una alerta paraneoplásica, ya que puede tratarse de un evento casual puesto que su observación es poco significativa:
- 1.
El penfigoide ampolloso (***) surge en edades avanzadas y la incidencia de neoplasias en estos pacientes no excede de la esperada en este grupo de edad, aunque se describen algunos casos más resistentes al tratamiento habitual acompañados de cáncer.
- 2.
Se admite el carácter paraneoplásico en algunos casos de penfigoide cicatricial (***), concretamente en la variante con anticuerpos antiepiligrina positivos.
- 3.
En la dermatitis herpetiforme (***), frecuentemente asociada a enteropatía por intolerancia al gluten, es conocido el posible desarrollo de linfoma intestinal en aquellos pacientes que tienen enfermedad celiaca.
- 4.
La dermatitis ampollosa IgA lineal (***) se ha descrito asociada a procesos linfoproliferativos y a carcinoma renal15–17.
- 5.
La epidermólisis ampollosa adquirida (***), caracterizada por anticuerpos anticolágeno tipo vii, puede ser expresión de tumores linforreticulares u otros18.
- 6.
De forma anecdótica se ha descrito un caso de pénfigo foliáceo (***) asociado a linfoma no Hodgkin que respondió a tratamiento con rituximab19.
- 7.
Al eritema exudativo multiforme (***) también podría atribuírsele un carácter paraneoplásico cuando es persistente y atípico en asociación con carcinoma renal, adenocarcinoma gástrico y colangiocarcinoma extrahepático20.
En este apartado se comentan dermatosis en las que predomina la presencia de pápulas, placas y nódulos a veces acompañados de inflamación. Algunas de ellas, como el xantogranuloma necrobiótico, la reticulohistiocitosis multicéntrica o el síndrome de Sweet tienen un carácter paraneolplásico acentuado. Otras, como el eritema nodoso o las lesiones de sarcoidosis, poseen menor relevancia en este campo21,22 (tabla 3).
Dermatosis con pápulas y nódulos inflamatorios con posible asociación a neoplasias
| Xantogranuloma necrobiótico** |
| Reticulohistiocitosis multicéntrica** |
| Síndrome de Sweet** |
| Pioderma gangrenoso** |
| Sarcoidosis cutánea*** |
| Eritema nodoso*** |
| Paniculitis necrotizante o necrosis grasa subcutánea** |
**Procesos ocasionalmente asociados a neoplasia interna; ***procesos excepcionalmente asociados a neoplasia interna.
Se caracteriza por placas múltiples amarillentas y nódulos subcutáneos distribuidos en la región periorbitaria, la cabeza, el cuello, las flexuras de las extremidades y el tronco. La ulceración y la cicatrización son frecuentes. En el 80% de los casos se asocia a gammapatía monoclonal benigna. Las asociaciones malignas incluyen procesos linfoproliferativos, mieloma, leucemia linfoide crónica y linfoma de Hodgkin y no Hodgkin3,23.
Reticulohistiocitosis multicéntrica (**)Se inicia con pápulas de pocos milímetros a 2cm de diámetro, de coloración normal o sonrosadas, marronáceas, localizadas en el dorso de los dedos alrededor del pliegue ungueal simulando un collarete. La erupción puede extenderse a la región periorbitaria, los codos, las rodillas, los pies, los hombros, etc. Histológicamente se aprecia un infiltrado histiocitario con células gigantes multinucleadas con citoplasma eosinófilo y aspecto vidrioso. Se acompaña de artropatía destructiva simétrica de manos y rodillas. El 20-25% de los pacientes afectados asocian malignidad hematológica, de mama, de estómago, de ovario y de cuello uterino3,23–26.
Síndrome de Sweet o dermatosis neutrofílica aguda febril (**)Es un proceso de aparición aguda que se inicia con una erupción cutánea de distribución asimétrica localizada en la cara, las extremidades y la parte superior del tronco. Consta de múltiples pápulas, placas induradas y nódulos inflamatorios dolorosos de tonalidad eritematosa o purpúrica, habitualmente acompañados de fiebre, neutrofilia y repercusión sistémica.
En la biopsia cutánea se observa un denso infiltrado neutrofílico localizado en la dermis papilar con un grado variable de edema sin vasculitis.
En un 10-40% de casos se le considera asociado a malignidad, a la que precede o es concomitante con ella. Fundamentalmente se trata de una leucemia mieloide aguda y otros procesos hematológicos. En menor proporción se evidencian tumores sólidos (carcinoma genitourinario, de mama, de pulmón y gastrointestinal). También puede cursar sin procesos malignos (idiopático, infecciones, medicamentos, etc.), pero hay una serie de rasgos que inducen a buscar una neoplasia subyacente: se trata de pacientes tanto de edad avanzada como niños que no tienen leucocitosis ni neutrofilia, pero sí anemia y trombopenia concomitantes; generalmente no presentan fiebre y las lesiones son dolorosas y localizadas en la cabeza, el cuello y las extremidades superiores, que a menudo desarrollan vesiculación, pústulas, ampollas hemorrágicas o ulceración y suelen afectarse las mucosas (figs. 3 y 4)3,27–31.
.")
Se sugiere que el pioderma gangrenoso y el síndrome de Sweet pueden formar parte del mismo espectro. De hecho el pioderma gangrenoso atípico (**), encuadrable en las dermatosis neutrofílicas ulceradas, se asocia a malignidades hematológicas similares a las del síndrome de Sweet con mayor frecuencia que el pioderma gangrenoso clásico. Se caracteriza por lesiones superficiales ampollosas que evolucionan a la formación de úlceras, localizadas en la cabeza y en el cuello fundamentalmente, aunque no se excluyen otras localizaciones (fig. 5)2,3,21,29.
Sarcoidosis cutánea (***)
Se manifiesta de forma muy variada con pápulas, placas, nódulos, lupus pernio, induración de cicatrices, ulceración, alopecia, hipopigmentación, lesiones liquenoides, psoriasiformes y más raramente con distrofia ungueal o con aspecto de ictiosis. Su asociación a malignidad se atribuye al desequilibrio inmunitario. Los trastornos hematológicos son los más frecuentes, pero también se describen una gran variedad de tumores sólidos asociados22.
Con menor frecuencia, otras lesiones inflamatorias como las del eritema nodoso (***), ponen de manifiesto la presencia de un linfoma de Hodgkin. Son lesiones atípicas, ya que persisten entre uno y 5 meses y no responden a los tratamientos habituales32.
Paniculitis necrotizante o necrosis grasa subcutánea (**)Esta entidad acompaña a procesos pancreáticos incluyendo carcinomas, por lo que también se denomina paniculitis pancreática. Se produce por la excesiva producción de lipasa pancreática que induce la necrosis grasa. Clínicamente se caracteriza por la presencia de placas dolorosas eritematosas localizadas en los glúteos y las regiones pretibiales, aunque pueden extenderse al tronco y a las extremidades superiores. Estas lesiones se ulceran, adquieren un aspecto abscesiforme y drenan al exterior un material marronáceo que traduce la licuefacción de la grasa subcutánea. En ocasiones surgen otros síntomas como fiebre, artralgias y eosinofilia. Su presencia obliga a descartar un carcinoma acinar de células pancreáticas33.
Xerosis, ictiosis, dermatitis exfoliativa (eritrodermia)La sequedad cutánea es un signo frecuente, a veces constitucional o forma parte de una dermatitis atópica. En algunas ocasiones pone de manifiesto una malignidad subyacente (tabla 4).
Procesos que cursan sequedad cutánea y posible asociación a neoplasias sistémicas
| Eczema craquelé o eczema esteatósico*** |
| Ictiosis adquirida** |
| Dermatitis exfoliativa generalizada/eritrodermia** |
**Procesos ocasionalmente asociados a neoplasia interna; ***procesos excepcionalmente asociados a neoplasia interna.
La xerosis o sequedad cutánea habitualmente se establece en personas de edad avanzada, en las extremidades inferiores en la zona pretibial y puede conducir en su grado extremo a un «eczema craquelé o eczema esteatósico» (***). Pacientes con procesos endocrinos y malnutrición se afectan con facilidad.
En el contexto maligno se describe asociada a linfomas, adenocarcinoma gástrico, glucagonoma o adenocarcinoma de páncreas34. En pacientes en que el eczema craquelé es más evidente en el tronco y las raíces de los miembros, con un aspecto más inflamatorio y resistente al tratamiento tópico esteroideo, es conveniente explorar adenopatías, hepatoesplenomegalia, realizar una analítica, radiografía de tórax y ecografía abdominal para el despistaje neoplásico35,36.
Un grado más del eczema craquelé conduce a la «Ictiosis adquirida» (**), que se presenta con pequeñas escamas romboidales blancas o marronáceas localizadas en las superficies extensoras de las extremidades y en el tronco. Las palmas, las plantas y las flexuras permanecen respetadas (fig. 6). El linfoma de Hodgkin es la causa fundamental en el contexto de malignidad, pero también se asocian otros procesos linfoproliferativos (micosis fungoide, reticulolinfosarcoma, mieloma múltiple) y no linfoproliferativos (disgerminoma de ovario, leiomiosarcoma, carcinoma de células transicionales del riñón, carcinoma hepatocelular, de mama, sarcoma de Kaposi). Una peculiaridad de la ictiosis adquirida es que suele seguir al diagnóstico del tumor en varias semanas o meses. Su patogenia se atribuye a una síntesis reducida de lípidos o a una respuesta inmune anómala3,37–39.

Por último, muchos pacientes desarrollan o comienzan con una «dermatitis exfoliativa generalizada» (**) de aspecto eritrodérmico y con intensa descamación. Es frecuente la presencia de alopecia, distrofia ungueal, adenopatías generalizadas, hipotermia, hipoalbuminemia e insuficiencia cardiaca. La mayoría corresponden a la invasión directa de un linfoma cutáneo de células T. Más raramente se asocia a otros linfomas, leucemias y tumores sólidos32,40.
Prurito y prúrigo (**)El prurito es un síntoma muy frecuente en Dermatología atribuible a múltiples procesos. Cuando la secuencia de entidades benignas se descarta, es obligado investigar una neoplasia como causa de prurito generalizado. Puede ser un signo inespecífico de malignidad, su patogenia es compleja y se implica la producción de mediadores como la histamina y la serotonina por parte de ciertos tumores y la disregulación de las células T2,23.
El linfoma de Hodgkin es el más frecuentemente asociado a prurito generalizado, ya que este síntoma subjetivo surge en más del 25% de los pacientes y es refractario a los tratamientos habituales32. También puede ser expresión de cáncer de mama, de síndrome carcinoide, de linfoma cutáneo de células T, de carcinomas gastrointestinales y hepatocelulares41.
El prurito generalizado intenso es un síntoma que puede acabar evidenciándose con lesiones de rascado y pápulas excoriadas en zonas accesibles (lesiones de prúrigo): tercio superior de la espalda, extremidades superiores e inferiores23,42.
Muy raramente el prurito localizado es síntoma de una malignidad subyacente. Un ejemplo es el prurito nasal asociado a tumor cerebral y supone un mal pronóstico2.
Hirsutismo e hipertricosisLa hipertricosis es el crecimiento de pelo en una cantidad y grosor excesivo en cualquier parte del cuerpo, y el hirsurtismo es el excesivo crecimiento de pelo y vello en la mujer con características y distribución masculina (tabla 5).
El hirsutismo (**) puede ser un proceso idiopático, de causa suprarrenal o hipofisaria. Cuando el inicio es abrupto a veces sugiere un tumor productor de andrógenos, habitualmente ovárico, y se acompaña de alteraciones hormonales y otros signos de virilización7.
Un proceso típicamente paraneoplásico y poco frecuente es la hipertricosis lanuginosa adquirida (*), que se desarrolla de forma brusca con pelo fino, no pigmentado, tipo lanugo, inicialmente localizado en la cara (fig. 7), se extiende al tronco, axilas y extremidades respetando las palmas, las plantas y los genitales. En las mujeres no se acompaña de otros signos de virilización. Su carácter paraneoplásico es muy acentuado. Es más frecuente en mujeres que en varones. Pueden tener además glositis dolorosa, queilitis angular e hipertrofia papilar de la lengua.
.")
En los varones se asocia a cáncer de pulmón seguido en frecuencia por el colorrectal. En las mujeres es más frecuente el colorrectal, después el de pulmón y el de mama. Pero se han descrito otras asociaciones tumorales: en ovario, útero, vejiga, páncreas, riñón, linfomas y leucemias. Puede preceder o seguir al diagnóstico del tumor, pero habitualmente es un signo de mal pronóstico23,29,43,44.
Hiperhidrosis y anhidrosis (***)La hiperhidrosis es un proceso de origen multifactorial y, aunque raramente, a veces subyace patología tumoral (tabla 6).
- 1.
La hiperhidrosis generalizada se observa en el feocromocitoma, tumor carcinoide, linfoma de Hodgkin (sudoración nocturna) y tumores de la corteza cerebral.
- 2.
La hiperhidrosis localizada craneofacial se produce en situaciones de estrés mínimo. Cuando es unilateral, afectando a una sola mejilla, desencadenada por la ingesta y acompañada de salivación se corresponde con el síndrome de Frey, producido por el daño de las fibras parasimpáticas del nervio auriculotemporal. Es la sudoración gustativa y surge en procesos parotídeos, a veces neoplásicos45,46.
- 3.
La hiperhidrosis segmentaria se produce por neoplasias intratorácicas que alteran el tronco del simpático (linfomas, mesoteliomas, tumor de Pancoast, carcinoma de pulmón contralateral y neurinoma mediastínico superior47). Se trata del síndrome del Arlequín, caracterizado por flushing y sudoración unilateral inducido por el calor y el ejercicio. También hay casos idiopáticos.
- 4.
La anhidrosis surge en tumores neurológicos que afectan al sistema nervioso central a nivel hipotalámico o de la médula espinal, o puede yuxtaponerse a la hiperhidrosis segmentaria.
Ciertos tumores benignos y algunos malignos aisladamente no son indicativos de malignidad, pero con frecuencia forman parte de síndromes complejos, algunos de ellos genodermatosis, que en su curso evolutivo tienen mayor incidencia de neoplasias6. De su conocimiento depende el diagnóstico y, por tanto, el tratamiento precoz que permite prolongar la supervivencia de estos pacientes (tabla 7).
- 1.
Las queratosis seborreicas son muy frecuentes en personas de edad avanzada, pero cuando surgen de forma brusca o sufren un aumento repentino en número y tamaño con prurito intenso se trata del signo de Leser-Trélat (**). Su carácter paraneoplásico permanece en eterno debate, puesto que ambos procesos son relativamente comunes en este grupo de edad. Este signo se emparenta con la acantosis nigricans maligna, a la que a veces acompaña48. Las lesiones pueden adquirir una distribución en árbol de Navidad y localizarse con mayor frecuencia en la espalda. El prurito es un dato importante. Los tumores a los que se asocia son adenocarcinomas gastrointestinales y procesos linfoproliferativos, con menor frecuencia a carcinoma de pulmón, de vejiga, renal, de ovario y de mama3,23,31,49.
- 2.
Un proceso similar al anterior, descrito en la raza negra, lo constituye la dermatosis eruptiva papulosa negra (**) que en alguna ocasión se ha visto asociada a malignidad50.
- 3.
La presencia de lesiones verrugosas múltiples distribuidas por el tronco, la cara y las extremidades, simulando verrugas víricas de crecimiento explosivo que confluyen y ocasionan a veces un aspecto grotesco al paciente, con deformidad de manos y pies, constituyen la papilomatosis florida cutánea (*). Es un proceso paraneoplásico que para algunos autores es una variante clínica de la acantosis nigricans maligna37. Se describe asociada a carcinoma gástrico, de mama, de pulmón y de ovario51–54.
- 4.
Los tumores múltiples de glándulas sebáceas (adenomas y carcinomas sebáceos) pueden formar parte del síndrome de Muir Torre (**), se localizan en el tronco, son familiares y se asocian a carcinomas viscerales del tracto gastrointestinal. Por el contrario, cuando son solitarios y localizados en la cabeza y en el cuello no suelen implicar malignidad29.
- 5.
La coincidencia de tricolemomas, lipomas y angiomas sugiere el síndrome de Cowden (**) (síndrome de hamartomas múltiples), proceso hereditario en que es posible el desarrollo de cáncer de mama, de tiroides, de endometrio, de pulmón y de colon29.
- 6.
La asociación de quistes epidérmicos múltiples y grandes, fibromas, leiomiomas, tricoepiteliomas y neurofibromas forma parte del síndrome de Gardner (**), proceso autosómico dominante que puede asociarse a cáncer de colon29.
- 7.
Fibrofoliculomas y tricosdiscomas se observan en el síndrome de Birt-Hogg-Dubé (**). Es un trastorno autosómico dominante en el que puede desarrollarse carcinoma de pulmón y renal29.
- 8.
Los leiomiomas múltiples distribuidos en banda o segmentarios (fig. 8) pueden asociarse a tumores renales y formar parte del síndrome hereditario leiomiomatosis/cáncer renal (**)29.
- 9.
Poco conocido es el síndrome familiar de nevus atípicos múltiples y cáncer pancreático y melanoma (***). Son familias con riesgo elevado de melanoma y cáncer pancreático29.
- 10.
En el síndrome de los neuromas mucosos múltiples (**), o síndrome de neoplasias endocrinas múltiples, se aprecian neuromas múltiples en los labios, en la lengua, en la mucosa labial y en las encías. Es un proceso hereditario autosómico dominante en el que se desarrollan con mayor frecuencia carcinoma medular de tiroides y feocromocitoma29.
- 11.
La neurofibromatosis tipo i (enfermedad de Von Recklinghausen) (**) es un síndrome neurocutáneo autosómico dominante que incluye moteado axilar e inguinal, manchas café con leche, neurofibromas y ocasionalmente neuromas plexiformes (fig. 9). Puede complicarse con degeneración maligna de los neuromas plexiformes. En su evolución pueden surgir neoplasias malignas como el tumor de Wilms, el rabdomiosarcoma, el retinoblastoma, el melanoma, el leiomiosarcoma intestinal, el meduloblastoma y la leucemia. Cuando se desarrollan feocromocitomas son benignos en el 90% de los casos. Se describe también una incidencia precoz familiar de cáncer de mama y otras malignidades ginecológicas2,29.
- 12.
La esclerosis tuberosa (**) es un trastorno genético autosómico dominante que condiciona la formación de hamartomas en la piel, el cerebro, el riñón y el corazón. Las lesiones cutáneas son características: angiofibromas faciales (fig. 10), fibromas periungueales (tumores de Koenen) (fig. 11), placas chagrin y máculas hipopigmentadas lanceoladas. Los tumores malignos que tienen mayor incidencia en estos pacientes son en primer lugar los astrocitomas cerebrales, seguidos de los carcinomas renales de células claras2.
- 13.
El síndrome de Gorlin o síndrome de los nevus basocelulares múltiples (**) es un proceso autosómico dominante que implica la presencia de carcinomas basocelulares múltiples, quistes mandibulares, pits palmoplantares y anomalías óseas, entre otros (figs. 12 y 13). Estos pacientes también pueden desarrollar otras malignidades como meduloblastomas, oligodendrogliomas, fibrosarcoma de ovario, linfoma de Hodgkin y no Hodgkin2.

.")
.")
.")
.")
.")
Tumores cutáneos con posible asociación a neoplasias internas
| Queratosis seborreicas (signo de Leser-Trélat)** |
| Dermatosis eruptiva papulosa negra** |
| Papilomatosis florida cutánea* |
| Tumores sebáceos (síndrome de Muir Torre)** |
| Tricolemomas, lipomas, angiomas (síndrome de Cowden)** |
| Quistes epidérmicos (Síndrome de Gardner)** |
| Fibrofoliculomas y tricodiscomas (Síndrome de Birt-Hogg-Dubé)** |
| Leiomiomas (síndrome hereditario leiomimatosis/cáncer renal) ** |
| Nevus atípicos (síndrome familiar de nevus atípicos múltiples, cáncer pancreático y melanoma)** |
| Neuromas múltiples (síndrome de los neuromas mucosos múltiples)** |
| Neurofibromas (neurofibromatosis tipo i)** |
| Angiofibromas (esclerosis tuberosa)** |
| Carcinomas basocelulares (síndrome de Gorlin o síndrome de los nevus basocelulares múltiples)** |
*Procesos genuinamente paraneoplásicos; **procesos ocasionalmente asociados a neoplasia interna.
En la boca se pueden observar múltiples manifestaciones en relación con varios de los signos paraneoplásicos que ya se han citado (tabla 8): la macroglosia (**) con amiloidosis sistémica y mieloma; las glositis (*) propias del eritema necrolítico migratorio y de la hipertricosis lanuginosa; las lesiones erosivas del pénfigo paraneoplásico (*); las proliferaciones verrugosas y papilomatosis en la lengua y en los labios (*) en la acantosis nigricans maligna y papilomatosis florida cutánea51; y ulceraciones orales (**) en el síndrome de Sweet55. La leucoplasia oral en la disqueratosis congénita56 y en la paquioniquia congénita57 se comentará en el apartado de «Alteraciones ungueales», que es el signo predominante en ambos procesos.
Alteraciones bucales observadas en neoplasias internas
| Macroglosia** |
| Glositis en hipertricosis lanuginosa y ENM* |
| Lesiones erosivas del pénfigo PN* |
| Proliferaciones verrugosas y papilomatosis en la lengua y en los labios de ANM* |
| Leucoplasia*** |
*Procesos genuinamente paraneoplásicos; **procesos ocasionalmente asociados a neoplasia interna; ***procesos excepcionalmente asociados a neoplasia interna. ANM: acantosis nigricans maligna; ENM: eritema necrolítico migratorio; PN: pénfigo paraneoplásico.
De forma aislada las alteraciones ungueales no suelen tener relevancia en las malignidades sistémicas, pero con frecuencia la distrofia de las uñas forma parte de otras entidades, algunas de ellas ya citadas con anterioridad (parte 1), como son la acroqueratosis paraneoplásica de Bazex (estriaciones horizontales o verticales, onicólisis y desestructuración total de la uña), la paquidermoperiostosis u osteopatía hipertrófica (acropaquia), el pénfigo paraneoplásico (paroniquia y distrofia ungueal tipo liquen), la esclerosis tuberosa (tumores de Koenen periungueales) y probablemente otros procesos en que la afectación ungueal surge de forma tardía (síndrome carcinoide, eritema gyratum repens, dermatitis exfoliativa generalizada, etc.).
Sin embargo, en algunos síndromes la distrofia ungueal constituye el signo fundamental. Se destacan los más relevantes en la tabla 9.
Es una rara entidad de etiología desconocida, aunque se sabe que la hipoplasia del sistema linfático desempeña un papel importante. En ocasiones es de carácter familiar. Se caracteriza por uñas de coloración amarillo-verdosa, muy curvadas, engrosadas con ausencia de cutícula y a veces con onicólisis (fig. 14). No es infrecuente que se detenga su crecimiento. Estas alteraciones ungueales pueden asociarse a un trastorno sistémico o cursar de forma aislada, pero conviene descartar procesos respiratorios, linfedema primario, enfermedades autoinmunes como la artritis reumatoide y tumores malignos como el cáncer de mama, linfomas, etc.58,59.
Disqueratosis congénita (**)
Es una enfermedad multisistémica recesiva ligada al cromosoma X. Desde el punto de vista cutáneo se caracteriza por la tríada de anomalías ungueales, hiperpigmentación reticulada y leucoplasia en cualquier mucosa, pero más frecuentemente en la oral.
Las alteraciones ungueales son precoces. Las uñas inicialmente son frágiles e irregulares, permanecen pequeñas y terminan siendo vestigios ungueales. Se afectan en principio las de las manos y con posterioridad las de los pies.
Estos pacientes tienen un alto riesgo de desarrollar anemia aplásica, síndrome mielodisplásico, leucemia y tumores sólidos. La leucoplasia facilita la mayor incidencia de carcinomas epidermoides en la boca, en el recto, en el cuello uterino, en la vagina, en el esófago y en la piel56.
Paquioniquia congénita (**)Se trata de un grupo de displasias ectodérmicas de herencia autosómica dominante. Existen dos tipos fundamentales, la paquioniquia congénita (PC) tipo 1 corresponde al síndrome de Jadassohn-Lewandosky y se acompaña de queratodermia palmoplantar, queratosis folicular de las rodillas y los codos y leucoqueratosis bucal que requiere estrecho seguimiento ante la posibilidad de desarrollo de carcinomas epidermoides. La PC tipo 2 (síndrome de Jakson-Lawler) cursa con esteatocitomas múltiples y quistes de pelo velloso, además de otras múltiples alteraciones.
Ambas se caracterizan por importante engrosamiento de las uñas secundario a una hiperqueratosis subungueal en la porción distal. Las uñas son de coloración amarillo-grisáceo con curvatura transversal marcada. La distrofia ungueal se desarrolla durante la infancia57.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



