

Las displasias ectodérmicas (DE) son un trastorno mayor hereditario raro que se caracteriza por cambios en las estructuras de origen ectodérmico. Dicho síndrome está caracterizado por ectrodactilia, hipoplasia de las glándulas mamarias y pezones, paladar leporino, estenosis del conducto lacrimal, hipoacusia, alteraciones urogenitales, displasia nasal, hipohidrosis, hipodontia, displasia gonadal y ausencia de piel o alteraciones pilosas1,2. Las causas posibles de la asimetría mamaria congénita pueden deberse a problemas evolutivos u hormonales, síndromes, e incluso trastornos familiares o genéticos. En este documento describimos a una familia con diferentes hallazgos clínicos a lo largo de cuatro generaciones.
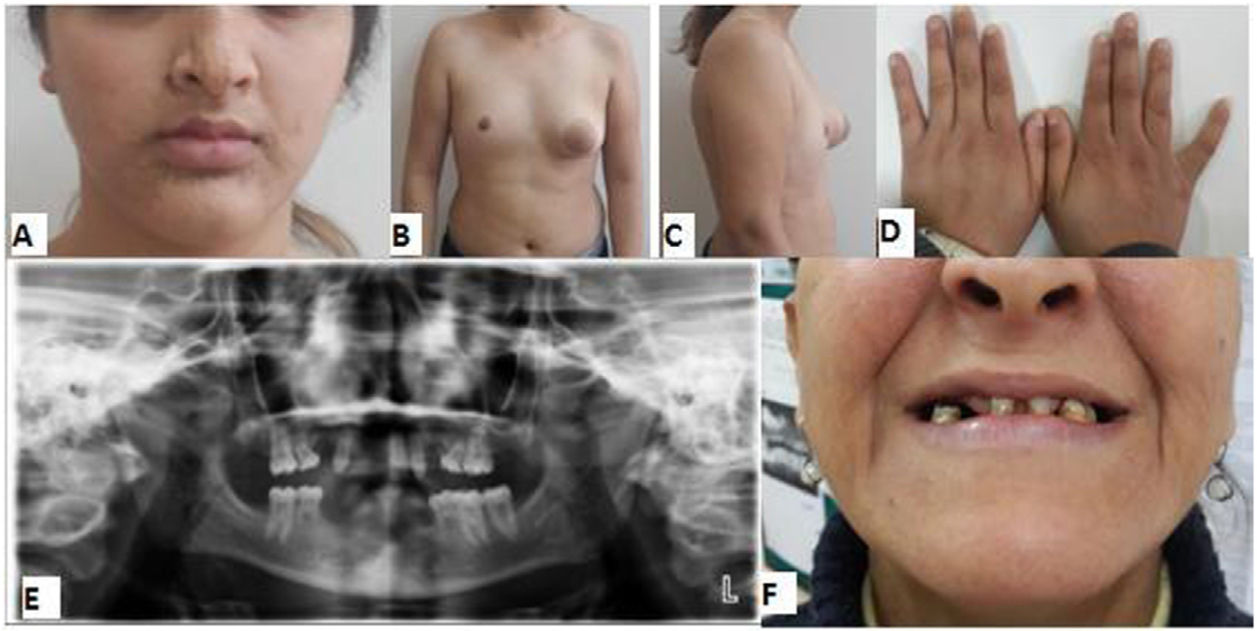
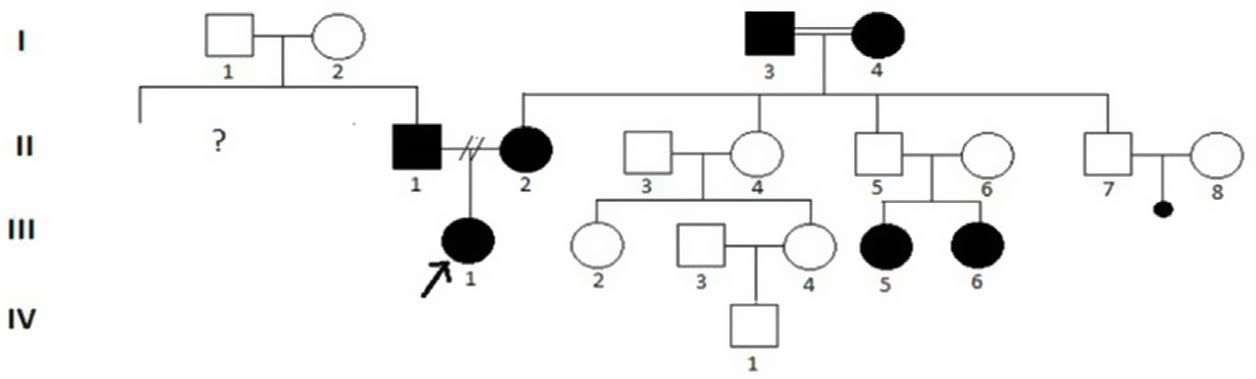
Una niña de 14 años acudió manifestando infradesarrollo de la mama derecha (fig. 1 B, C). La exploración clínica reveló pezón normal, aunque el tejido mamario izquierdo era anormal (E-5), y ausencia de areola en el aspecto profundo del pezón. El músculo pectoral mayor derecho era normal. También tenía parálisis facial (fig. 1 A), áreas cutáneas ampliamente despigmentadas, hiperpigmentación y dientes irregulares y ausentes (fig. 1 E), hipoacusia, pelo púbico P4-5 y pelo axilar ligeramente elongado (A-2). Los genitales externos, útero y ovarios eran normales, pero no existía clitoromegalia. También tenía las manos pequeñas (fig. 1 D), cerramiento de la epífisis, atrofia en el riñón izquierdo y problemas endocrinológicos. Los valores de las hormonas tales como FSH, LH, E2, prolactina, progesterona, testosterona, TSH y FT4 y el desarrollo mental y motor eran normales. El padre y la madre tenían diagnóstico de trastorno obsesivo compulsivo y retraso mental leve. Se observaron en ellos problemas dentales físicos (fig. 1 F) y trastornos del lenguaje. También se reportó que las glándulas sudoríparas de la madre habían perdido su función durante la infancia, tenía bocio, problemas alérgicos y dificultades de aprendizaje, y el cabello del padre no había crecido y era anormal. En los abuelos de la primera generación se registraron cosanguinidad (I-3,4), retraso mental y en el aprendizaje leves, y trastornos nerviosos y biliares. Sin embargo, de igual forma se encontró hipoacusia congénita en otros dos miembros de la tercera generación (primos, III-5,6) (fig. 2).
 (B, C), pequeño pezón izquierdo (B, C), parálisis facial (A), manos pequeñas (D), ausencia de dientes (E) y dientes de la madre (F).")

Se determinó que nueve dientes permanentes de la paciente seguían en fase de crecimiento, estando todavía en su lugar cuatro de los primarios. Se registró que los permanentes fueron los incisivos centrales anteriores superiores izquierdos, los caninos derecho e izquierdo superiores, los primeros molares derecho e izquierdo superiores, los primeros molares inferior derecho e inferior izquierdo y los segundos molares superiores izquierdos. Los caninos tenían características dismorfológicas tales como que la forma de los incisivos era ungueal y se afilaba hacia los bordes; el resto de los dientes permanentes tenían características morfológicas normales. No se detectaron dientes retenidos ni pérdidas dentales. Se observó un molar deciduo en cada mitad de la mandíbula, así como pérdida ósea en la parte anterior inferior debido a edentulismo (fig. 1 E).
Los resultados citogenéticos de nuestra paciente reflejaron un complemento cromosómico normal. Sin embargo, se encontraron cambios cromosómicos estructurales y numéricos en 22,5% de las células de la madre.
Los hallazgos clínicos y fenotípicos de los padres de la paciente también fueron consistentes con DE. Además, se reportó que la madre y el padre de la madre (abuelos, I-3,4) eran cosanguíneos y tenían hallazgos clínicos similares. De hecho, se registró que las dos hijas del tío de la probando tenían sordera congénita (primas, III-5,6). Los antecedentes familiares nos muestran que los hallazgos clínicos de la paciente son hereditarios. Se han constatado anteriormente casos de asimetría mamaria en algunas familias. En una de ellas, se notificó ausencia de mama bilateral en un varón, tres de entre cuatro jóvenes y tres nietos de tres generaciones3. Por otro lado, en un caso en el que la madre y el padre no estaban afectados, los primos y primas en otra familia no tenían mamas bilaterales, reportándose que siete individuos de cuatro generaciones carecían de mamas o eran hipoplásicas4,5. Nuestros hallazgos sugieren que la presencia de mamas asimétricas es una patología grave y rara de DE.
Dado que la cápsula ótica es de origen ectodérmico, es natural ver hipoacusia sensorineural en este tipo de displasia. En la familia actual, tres miembros de la generación (la probando y las primas III-1,5,6) tenían hipoacusia congénita, la cual fue consistente con series de casos previos6. Se observó que el desarrollo motor, cognitivo y de inteligencia estaban afectados en otros miembros de la familia. Algunos estudios han reportado un desarrollo motor/mental anormal en 15–25% de los pacientes afectados de DEH7. También se han identificado diversos problemas renales. Nosotros reportamos estructura renal atrófica en la paciente. Su madre tenía muchas irregularidades estructurales y numéricas en diferentes cromosomas. Debe considerarse que los daños en el cromosoma 2 (regiones q11-ter, q14, q21, q23 y p23), 19, 21 y demás pérdidas cromosómicas detectadas en ella pueden estar asociadas al desarrollo del tejido ectodérmico. Un gen responsable de DEH autosómica ha sido recientemente mapeado con el cromosoma 2q11-q13 en humanos y ratones8. También ha habido expresión de dicho en el epitelio y en las capas adyacentes o parcialmente suprayacentes de la piel humana en desarrollo9. Toda esta información muestra que el cromosoma 2 juega un papel en la etiología de DE.
Conflicto de interesesLos autores declaran la ausencia de conflicto de intereses.
Quisiéramos agradecer al Departamento de Pediatría la derivación de la familia a nuestro departamento. Quisiera expresar mi gratitud a la tía, quien cuidó estrechamente a la paciente índice y a su madre, contribuyó a la adquisición de la información y completó el formulario de consentimiento en concordancia con la aprobación de ambas.



