







Birt–Hogg–Dubé syndrome is a rare autosomal dominant genodermatosis that is characterized by the presence of fibrofolliculomas and/or trichodiscomas, pulmonary cysts, spontaneous pneumothorax, and renal tumors. The most common histological types found in renal tumors from patients with the syndrome are oncocytoma–chromophobe carcinoma hybrids and pure forms of chromophobe carcinoma, oncocytic carcinoma, and clear cell or papillary cell carcinoma. The syndrome is linked to mutations in the FLCN gene, which encodes folliculin and is preferentially expressed in the skin, kidney, and lung. The syndrome can exhibit a high degree of clinical variability, and the skin lesions that are a warning sign for dermatologists may be absent in up to 70% of cases. Consequently, although skin lesions and mutations in FLCN are the main diagnostic criteria for Birt–Hogg–Dubé syndrome, a diagnosis can be made based on noncutaneous manifestations, with or without known family history of the syndrome, even in the absence of histological confirmation of fibrofolliculomas or trichodiscomas.
El síndrome de Birt–Hogg–Dubé (SBHD) es una rara genodermatosis de herencia autosómica dominante caracterizada esencialmente por la presencia de fibrofoliculomas y/o tricodiscomas, quistes pulmonares, neumotórax espontáneos y cánceres renales, siendo los tipos histológicos más frecuentes las formas híbridas de oncocitoma y carcinoma cromófobo o formas puras de carcinoma cromófobo, oncocítico, de células claras o papilar. El gen implicado en este síndrome, FLCN, codifica la foliculina, que se expresa preferentemente a nivel cutáneo, renal y pulmonar. Este síndrome puede presentarse con una gran variabilidad clínica, y las lesiones cutáneas que son el signo de alarma para los dermatólogos pueden estar ausentes hasta en un 70% de los casos. Así, aunque las lesiones cutáneas son, junto con las mutaciones del gen FLCN, los criterios mayores para el diagnóstico del SBHD, este diagnóstico es posible incluso cuando no existe confirmación histológica de fibrofoliculomas o tricodiscomas, por las mencionadas manifestaciones extracutáneas, con o sin antecedentes familiares conocidos.
In 1977, Birt et al.1 described a series of patients with multiple papular skin-colored, dome-shaped lesions located on the face, neck, and trunk. Histologically, these lesions corresponded to fibrofolliculomas, trichodiscomas, and soft fibromas. This was the first description of what later came to be known as Birt–Hogg–Dubé syndrome, a genodermatosis that exhibits autosomal dominant inheritance. More than 20 years later, its association with renal carcinoma and/or pulmonary lesions was discovered.2
Clinical FeaturesSkin ManifestationsThe skin manifestations of Birt–Hogg–Dubé syndrome essentially comprise fibrofolliculomas and/or trichodiscomas that usually appear in individuals in their 20s or 30s. Trichofolliculomas, trichodiscomas, and acrochordons—traditionally considered as the characteristic triad of Birt–Hogg–Dubé syndrome—appear to belong to the histopathologic and clinical spectrum of fibrofolliculoma. Clinically, these lesions are practically indistinguishable from one another and present as multiple whitish papules, with a dome-shaped appearance, a few millimeters in diameter, located mainly on the nose, forehead, and cheeks, although they can also appear on the neck and trunk (Fig. 1).3 Histologically, fibrofolliculoma is typically vertical or perpendicular to the epidermis, as it forms around a hair follicle. It is characterized by the presence of long, thin, mantle-like epithelial cords, which sometimes terminate in mature sebaceous glands, with surrounding fibrovascular stroma containing a variable myxoid component (Fig. 2). Unlike fibrofolliculomas, trichodiscomas are oriented horizontally, that is, parallel to the epidermis, and are composed exclusively of stroma identical to that of fibrofolliculomas, surrounded at the base by hair follicle units (Fig. 3). Trichodiscoma is considered a late form of fibrofolliculoma.4

.")
.")
In addition to these characteristic lesions, the presence of facial angiofibromas has occasionally been reported, as has the involvement of oral mucosa in the form of multiple papules on the lips and buccal and gingival mucosa.5 The fibrofolliculomas and trichodiscomas found in Birt–Hogg–Dubé syndrome have features in common with the angiofibromas that occur in tuberous sclerosis complex. Histologically, the presence of a stroma with thicker collagen bundles, along with stellate fibroblasts, as well as the absence of the epithelial component, are useful features for differentiating angiofibromas from fibrofolliculomas. Analysis of immunoreactivity for CK15, CD34, and factor XIIIa is not very useful as it may occur in both types of lesion. In Birt–Hogg–Dubé syndrome, a mutation in the folliculin gene (FCLN) is associated with the development of multiple fibrofolliculomas and trichodiscomas, whereas mutations in TS1 and TS2 are linked to the appearance of angiofibromas in the tuberous sclerosis complex. Recent work has shown that both proteins could form part of the same mammalian target of rapamycin (mTOR) pathway, responsible for the development of the characteristic skin lesions in both syndromes.4 In fact, the presence of fibrofolliculomas in patients with tuberous sclerosis or of angiofibromas in patients with Birt–Hogg–Dubé syndrome has been very occasionally reported.5 In addition, patients with Birt–Hogg–Dubé syndrome and those with tuberous sclerosis can present with lung and renal disease. These similarities in clinical manifestations, and the common histologic features in skin lesions, are further evidence that genetic abnormalities arise in a common pathway. In fact, the results of a study conducted in Schizosaccharomyces pombe, a unicellular eukaryotic yeast, suggest that the genes responsible for Birt–Hogg–Dubé syndrome and tuberous sclerosis both act via the mTOR pathway and lead to renal cell carcinoma, skin cancer, and pulmonary symptoms through mechanisms involving inappropriate inhibition in Birt–Hogg–Dubé syndrome and activation in tuberous sclerosis.6
Extracutaneous ManifestationsIn addition to skin lesions, patients with Birt–Hogg–Dubé syndrome may present a series of extracutaneous lesions.7 In recent years, it has been shown that these patients are at a greater risk of developing renal cancer and spontaneous pneumothorax.8–11 In 1993, Roth et al.9 described the first case of renal cancer in a patient with Birt–Hogg–Dubé syndrome. Toro et al.2 subsequently described Birt–Hogg–Dubé syndrome as a marker of renal cancer. Further studies providing confirmation have since been published.7,11–14 It was reported that patients with Birt–Hogg–Dubé had a 7-fold higher risk of renal cancer, with a predilection for men and an age of onset between 20 and 55 years.14 Seven different autosomal dominant syndromes have been identified as having an association with renal cancer.15 However, unlike the other syndromes, the presentation of renal lesions can be bilateral and multifocal in the case of Birt–Hogg–Dubé syndrome, with 5 different histologic types: oncocytic–chromophobe hybrid carcinoma (50%), purely chromophobe carcinomas (34%), purely oncocytic carcinomas (5%), clear cell carcinomas (3%), and papillary carcinomas (2%).11,14 The prevalence of renal tumors in patients with germline mutations in the FLCN gene varies from 6.5% to 34% according to different studies.11 With regard to the risk of metastasis, to date, only 5 cases of distant metastases of renal cancer have been reported.11 Clinicopathologic studies showed that the renal tumors of these patients were of clear-cell, tubulopapillary, or papillary subtypes, which account for a minority of subtypes in Birt–Hogg–Dubé syndrome.
Up to 80% of patients with Birt–Hogg–Dubé syndrome have lung cysts, which may be asymptomatic for years.9,11,14–16 The number and size of the lesions varies from patient to patient, ranging from small cysts to bullae measuring several centimeters across, located mainly in the lung bases and at the subpleural level9 (Fig. 4). A relationship between size and volume of the cysts and the risk of pneumothorax has been reported—the larger the volume the greater the risk. The mean age of presentation is 38 years, with no clear predilection for either sex. Some studies suggest that male sex is a risk factor for spontaneous pneumothorax,17 whereas others report no such risk.9 Among patients who present lung cysts, approximately 20–30% have a history of pneumothorax, with a mean of 2 prior episodes.9 In addition, when patients with a history of pneumothorax are studied, the vast majority have multiple cysts. The right lung is more frequently affected, although both lungs can be involved in up to 23% of cases.9 In patients with Birt–Hogg–Dubé syndrome, the risk of pneumothorax is 50 times greater than in the normal population.16 The most frequent mutation in patients with a history of pneumothorax or lung lesions and Birt–Hogg–Dubé syndrome is c.1733ins/delC located in exon 119. Patients with certain mutations are at risk of developing larger and more numerous cystic lesions. Thus, the mutation in exon 9 confers a risk of developing a greater number of cysts, whereas mutations in exon 9 and 12 are associated with larger cysts.9 The pathophysiology of lung cysts is unknown but it may be that haploinsufficiency is enough to induce the development of these lesions, as is the case in skin tumors. In addition to the characteristic associations described above, in patients with Birt–Hogg–Dubé syndrome, there have been reports of adenomas and colorectal polyps, parotid oncocytomas, parathyroid oncocytomas, neural tumors, trichoblastomas, lipomas, angiolipomas, connective tissue abnormalities, chorioretinopathy, and nonrenal malignant neoplasms such as breast cancer, colorectal cancer, tonsil cancer, and lung cancer, and also skin cancers such as melanoma, basal cell carcinoma, squamous cell carcinoma, dermatofibrosarcoma, and leiomyosarcoma.18 Despite these reports, a significant association between Birt–Hogg–Dubé syndrome and these neoplasms has not yet been demonstrated, and further studies are needed.19 Some authors maintain that carriers of the c.1285dupC mutation have a greater risk of colon cancer.19
Genetic Basis Plain chest X-ray showing left spontaneous pneumothorax in a patient with Birt–Hogg–Dubé syndrome. (B) Chest computed tomography image showing multiple right lung cysts in a patient with the syndrome.")
In 2002, the FLCN gene was identified as being associated with Birt–Hogg–Dubé syndrome.12 This gene, located on chromosome 17p11.2 and comprising 14 exons, encodes a 64kDa protein, folliculin, which is expressed in most tissues including the skin and its appendages, the lungs (type 1 pneumocytes) and the kidney (distal nephron). The exact function of this protein has not yet been elucidated, but it seems to be implicated in the adenosine-monophosphate-activated protein kinase and mTOR pathways.13,20
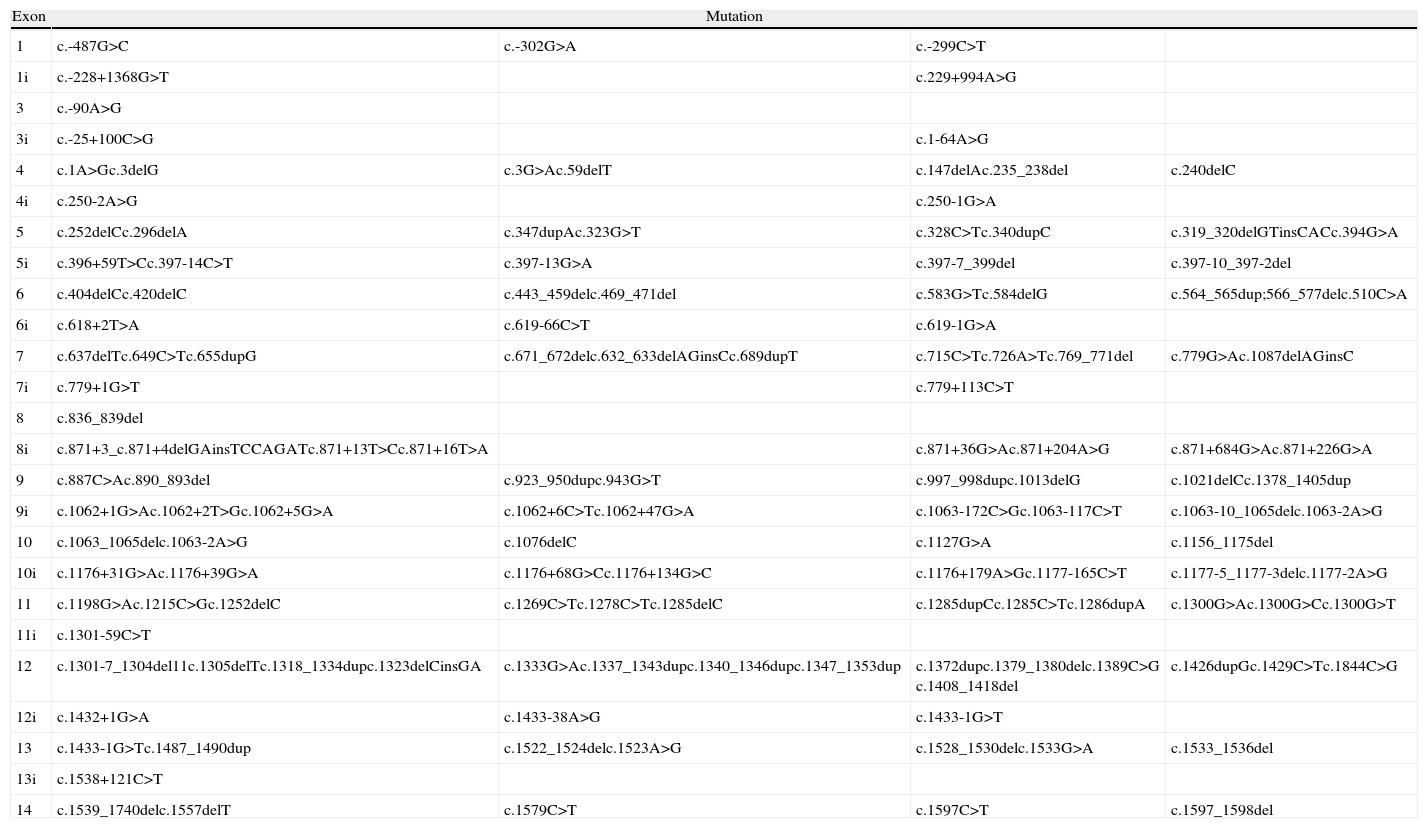
Although mutations can occur anywhere in the FLCN gene, including exons and introns,21 to date 50% of the mutations reported are frameshifts caused by insertions or deletions at the mutational hotspot in the cytosine 8 nucleotide of exon 11.21,22 Mutations 1285dupC (previously described as c.1733insC, c.1740dupC, or c.1277insC) and c.1285delC (also described as c.1733delC) are the most frequently reported mutations in patients with Birt–Hogg–Dubé syndrome and their family members. The impact on protein transcription depends on the type of mutation (base substitution, nucleotide deletion or insertion, or interference in messenger RNA splicing). Table 1 presents the mutations described to date in the FLCN gene.12,16,21–24 The loss of FLCN expression observed in almost all histologic subtypes of renal carcinoma implicates this gene in the pathogenesis of this type of tumor and provides further support for its function as a tumor suppressor gene.24 A second somatic mutation is necessary for renal cancer in patients with Birt–Hogg–Dubé syndrome to develop; this mutation leads to loss of mRNA expression of FLCN in these tumors.12,15,23
Germline Mutations in Birt–Hogg–Dubé Syndrome.
| Exon | Mutation | |||
| 1 | c.-487G>C | c.-302G>A | c.-299C>T | |
| 1i | c.-228+1368G>T | c.229+994A>G | ||
| 3 | c.-90A>G | |||
| 3i | c.-25+100C>G | c.1-64A>G | ||
| 4 | c.1A>Gc.3delG | c.3G>Ac.59delT | c.147delAc.235_238del | c.240delC |
| 4i | c.250-2A>G | c.250-1G>A | ||
| 5 | c.252delCc.296delA | c.347dupAc.323G>T | c.328C>Tc.340dupC | c.319_320delGTinsCACc.394G>A |
| 5i | c.396+59T>Cc.397-14C>T | c.397-13G>A | c.397-7_399del | c.397-10_397-2del |
| 6 | c.404delCc.420delC | c.443_459delc.469_471del | c.583G>Tc.584delG | c.564_565dup;566_577delc.510C>A |
| 6i | c.618+2T>A | c.619-66C>T | c.619-1G>A | |
| 7 | c.637delTc.649C>Tc.655dupG | c.671_672delc.632_633delAGinsCc.689dupT | c.715C>Tc.726A>Tc.769_771del | c.779G>Ac.1087delAGinsC |
| 7i | c.779+1G>T | c.779+113C>T | ||
| 8 | c.836_839del | |||
| 8i | c.871+3_c.871+4delGAinsTCCAGATc.871+13T>Cc.871+16T>A | c.871+36G>Ac.871+204A>G | c.871+684G>Ac.871+226G>A | |
| 9 | c.887C>Ac.890_893del | c.923_950dupc.943G>T | c.997_998dupc.1013delG | c.1021delCc.1378_1405dup |
| 9i | c.1062+1G>Ac.1062+2T>Gc.1062+5G>A | c.1062+6C>Tc.1062+47G>A | c.1063-172C>Gc.1063-117C>T | c.1063-10_1065delc.1063-2A>G |
| 10 | c.1063_1065delc.1063-2A>G | c.1076delC | c.1127G>A | c.1156_1175del |
| 10i | c.1176+31G>Ac.1176+39G>A | c.1176+68G>Cc.1176+134G>C | c.1176+179A>Gc.1177-165C>T | c.1177-5_1177-3delc.1177-2A>G |
| 11 | c.1198G>Ac.1215C>Gc.1252delC | c.1269C>Tc.1278C>Tc.1285delC | c.1285dupCc.1285C>Tc.1286dupA | c.1300G>Ac.1300G>Cc.1300G>T |
| 11i | c.1301-59C>T | |||
| 12 | c.1301-7_1304del11c.1305delTc.1318_1334dupc.1323delCinsGA | c.1333G>Ac.1337_1343dupc.1340_1346dupc.1347_1353dup | c.1372dupc.1379_1380delc.1389C>G c.1408_1418del | c.1426dupGc.1429C>Tc.1844C>G |
| 12i | c.1432+1G>A | c.1433-38A>G | c.1433-1G>T | |
| 13 | c.1433-1G>Tc.1487_1490dup | c.1522_1524delc.1523A>G | c.1528_1530delc.1533G>A | c.1533_1536del |
| 13i | c.1538+121C>T | |||
| 14 | c.1539_1740delc.1557delT | c.1579C>T | c.1597C>T | c.1597_1598del |
Contrary to what occurs in renal tumors, no loss of heterozygosity has been detected in skin lesions and the level of FLCN mRNA expression is high, although the reason for this is not known.24 These findings suggest that different mechanisms are implicated in the development of these skin tumors and renal cancers. Further studies are therefore required to clarify the mechanisms implicated in skin lesions.
To date, no correlation between genotype and phenotype has been established, and further studies are needed. However, some authors have observed a greater frequency of mutations in exon 11 in those patients with a history of pneumothorax, as well as a relationship between mutations in exon 9 and 12 and the number and size, respectively, of lung cysts.9 Other authors suggest that patients with the c.1285delC mutation have a lower risk of developing renal cancer, but further studies are necessary.11
DiagnosisTable 2 shows the current diagnostic criteria proposed by the European Birt–Hogg–Dubé Consortium.25 Of note is that, although skin lesions are a warning sign for dermatologists, they are not present in all patients with Birt–Hogg–Dubé syndrome. In fact, not all patients present with the traditional triad of skin, renal, and lung disease.16 Kunogi et al.16 observed that up to 70% of patients with Birt–Hogg–Dubé syndrome and pneumothorax did not present with skin lesions or renal disorders. Thus, given the clinical variability of this syndrome, it should be suspected not only in patients with characteristic skin lesions but also in those with extracutaneous lesions as described above.
Criteria Proposed by the European Birt–Hogg–Dubé Consortium.
| Major Criteria |
| At least 5 fibrofolliculomas or trichodiscomas, at least 1 confirmed histologically, of adult onset |
| Pathogenic germline mutation in FLCN |
| Minor Criteria |
| Multiple lung cysts: basally located with no other apparent cause, with or without spontaneous pneumothorax |
| Renal cancer: early onset (<50 years), multifocal or bilateral renal cancer, or renal cancer of characteristic histologic forms (oncocytic-chromophobe hybrid histology) |
| First-degree relative with Birt–Hogg–Dubé syndrome |
Patients should fulfill 1 major or 2 minor criteria for diagnosis of Birt–Hogg–Dubé syndrome.
The European Birt–Hogg–Dubé Consortium has proposed a series of criteria that represent an indication for genetic study. These include early-onset renal carcinoma (<50 years), particularly if multifocal or bilateral and of characteristic histology (chromophobe, oncocytic, or hybrid), and when unexplained cystic lung disease or spontaneous pneumothorax is present, especially in the case of bilateral disease and cysts located at the base of the lung.25 In addition, those patients with a family history of pulmonary cystic disease, spontaneous pneumothorax, renal carcinoma, or a combination of these conditions should also be considered as candidates for genetic study (Table 3).25 Thus, in those patients whose only manifestation is lung or renal involvement, detection of germline mutation in FLCN would help confirm diagnosis and allow genetic counseling. Genetic counseling units are available in almost all autonomous regions of Spain, and these can be consulted for requesting the genetic studies.26
Criteria for Requesting Genetic Study According to the European Birt–Hogg–Dubé Consortium.
| Characteristic skin lesions (fibrofolliculomas and/or trichodiscomas) |
| Cystic lung disease with no apparent cause |
| Spontaneous primary pneumothorax |
| Renal cancer: early onset (<50 years), multifocal or bilateral renal cancer, or renal cancer of characteristic histologic forms (pure or oncocytic–chromophobe hybrid histology) |
| First-degree relative with any of the above characteristics |
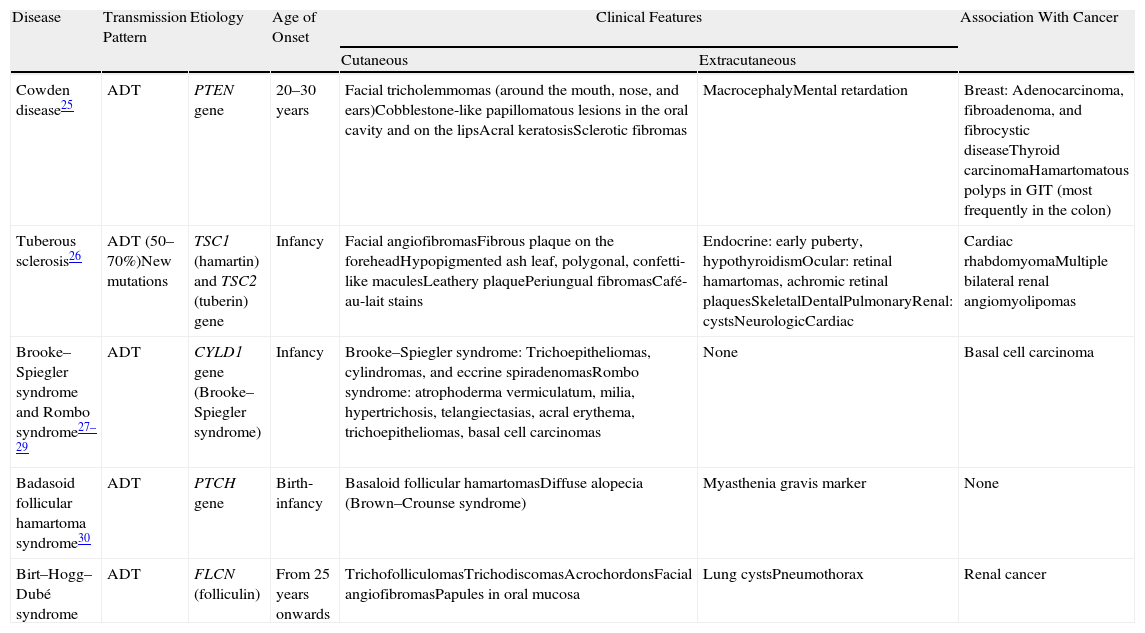
Differential diagnosis in a patient with papular facial lesions should include other syndromes such as Cowden syndrome, Brooke–Spiegler syndrome, Rombo syndrome, tuberous sclerosis, and basaloid follicular hamartoma syndrome.25,27–33 Diagnosis is based on clinical features and histologic study. Table 4 shows the principal features of each of these syndromes.
Differential Diagnosis in a Patient With Facial Papular Lesions.
| Disease | Transmission Pattern | Etiology | Age of Onset | Clinical Features | Association With Cancer | |
| Cutaneous | Extracutaneous | |||||
| Cowden disease25 | ADT | PTEN gene | 20–30 years | Facial tricholemmomas (around the mouth, nose, and ears)Cobblestone-like papillomatous lesions in the oral cavity and on the lipsAcral keratosisSclerotic fibromas | MacrocephalyMental retardation | Breast: Adenocarcinoma, fibroadenoma, and fibrocystic diseaseThyroid carcinomaHamartomatous polyps in GIT (most frequently in the colon) |
| Tuberous sclerosis26 | ADT (50–70%)New mutations | TSC1 (hamartin) and TSC2 (tuberin) gene | Infancy | Facial angiofibromasFibrous plaque on the foreheadHypopigmented ash leaf, polygonal, confetti-like maculesLeathery plaquePeriungual fibromasCafé-au-lait stains | Endocrine: early puberty, hypothyroidismOcular: retinal hamartomas, achromic retinal plaquesSkeletalDentalPulmonaryRenal: cystsNeurologicCardiac | Cardiac rhabdomyomaMultiple bilateral renal angiomyolipomas |
| Brooke–Spiegler syndrome and Rombo syndrome27–29 | ADT | CYLD1 gene (Brooke–Spiegler syndrome) | Infancy | Brooke–Spiegler syndrome: Trichoepitheliomas, cylindromas, and eccrine spiradenomasRombo syndrome: atrophoderma vermiculatum, milia, hypertrichosis, telangiectasias, acral erythema, trichoepitheliomas, basal cell carcinomas | None | Basal cell carcinoma |
| Badasoid follicular hamartoma syndrome30 | ADT | PTCH gene | Birth-infancy | Basaloid follicular hamartomasDiffuse alopecia (Brown–Crounse syndrome) | Myasthenia gravis marker | None |
| Birt–Hogg–Dubé syndrome | ADT | FLCN (folliculin) | From 25 years onwards | TrichofolliculomasTrichodiscomasAcrochordonsFacial angiofibromasPapules in oral mucosa | Lung cystsPneumothorax | Renal cancer |
Abbreviations: ADT, autosomal dominant transmission; GIT, gastrointestinal tract.
Familial spontaneous pneumothorax may occur in different hereditary processes. Thus, when faced with a patient with a family history of spontaneous pneumothorax and lung cysts, the differential diagnosis should include other entities that may present with cystic changes in the lung parenchymya.9 These include α1-antitrypsin deficiency, Marfan syndrome, Ehlers–Danlos syndrome, lymphangioleiomyomatosis, tuberous sclerosis, Langerhans cell histiocytosis, and cystic fibrosis. Idiopathic pulmonary fibrosis, Pneumocystis jiroveci infection, lymphocytic interstitial pneumonia, and septic embolism should also be included in the differential diagnosis. The site of the lung cysts, a family history of such cysts, and the presence of other accompanying lesions, particularly cutaneous ones, can help guide the diagnosis.
Management of Patients With Birt–Hogg–Dubé SyndromeAs we have seen, the clinical manifestations of Birt–Hogg–Dubé syndrome are not just limited to skin lesions, and other body systems can be involved. The management of these patients should therefore be multidisciplinary and involve the cooperation of different specialists. Diagnosis of skin lesions associated with this syndrome is based on clinical and histologic features. An appropriate skin examination along with excision of a suspect lesion is necessary. Treatment should be considered on esthetic grounds. In addition to surgery, the use of oral isotretinoin and treatment with carbon dioxide lasers and erbium:YAG lasers have been reported.27,34,35 Laser ablation is a useful treatment for other adnexal tumors. Gambichler et al.34 described treatment of a patient with Birt–Hogg–Dubé syndrome and skin lesions with an erbium:YAG laser (spot size 1.5mm, fluence 5J/cm2, pulse energy 340mJ). Subsequently, in 2001, Jacob and Dover35 used a combination of carbon dioxide and erbium:YAG lasers to treat a patient with similar lesions. Currently, van Steensel36 is conducting a double-blind, phase III study to assess the efficacy of topical rapamycin (0.25mL of rapamycin, 1mg/mL oral solution, administered twice daily) in the management of these lesions. The results of this study will be available in the near future.
As commented earlier, patients with Birt–Hogg–Dubé syndrome have a higher risk of developing renal cancer. Those patients who are carriers of FLCN germline mutations and those patients who are at risk should be monitored closely. There are no established guidelines for defining at which age to start, the best screening approach, or the frequency of follow-up visits. As the age of onset of renal cancer is between 25 and 75 years, the age for initiation of follow-up could be set at 20 years.27 Ultrasound, computed tomography (CT), and magnetic resonance imaging may be useful imaging techniques. Some authors suggest using renal ultrasound and/or abdominal CT on diagnosis and then every 3 or 5 years.18 Staging in the event of diagnosis of renal cancer is no different to renal cancer in patients without the syndrome. Once diagnosed, treatment is usually surgical, leaving as much renal parenchyma as possible with nephron-sparing techniques instead of radical nephrectomy.37 In the event of metastases, rapamycin analogs may be considered, given the implication of the FLCN gene in regulating the mTOR pathway, although more studies are needed to demonstrate benefit.27
With regard to the management of lung disease, patients with Birt–Hogg–Dubé syndrome may have a greater risk of pneumothorax when subjected to pressure changes, for example, during journeys in airplanes or aquatic activities such as diving. It is not clear to what extent patients should be advised against undertaking such activities, but the patient should be informed of the risk, and also educated about the signs and symptoms that may appear. There are insufficient data to support a clear association with smoking, but a greater prevalence of renal cancer and pneumothorax has been observed in smokers, and so patients should be advised to give up tobacco use.9 Patients with a history of recurring pneumothorax or symptomatic lung disease should be referred to a lung specialist. Chest X-ray or CT is useful for diagnosis of lung cysts, but there are no universally accepted protocols for monitoring these lesions. Treatment is similar to those patients who present with spontaneous pneumothorax not associated with the syndrome.
With regard to the association between Birt–Hogg–Dubé syndrome and colon cancer, no specific indication has been reported for colonoscopy in these patients, and the recommended management is the same as in the general population.19 Some authors suggest that this association could be mere coincidence given the frequency of colon cancer in the general population.19,38
ConclusionIn conclusion, even though the warning signs for dermatologists are skin lesions with a histologically confirmed diagnosis such as fibrofolliculomas and trichodiscomas, these are often absent. Extracutaneous manifestations such as recurrent spontaneous pneumothorax or basal lung cysts, as well as certain renal cancers, along with genetic study, can be used to establish a diagnosis of Birt–Hogg–Dubé syndrome.
FundingThis study was performed with the support of a grant (AP-088/11) for health research projects, awarded by the Department of Health of the Autonomous Community of Valencia.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: López V, et al. Actualización en el síndrome Birt–Hogg–Dubé. Actas Dermosifiliogr. 2012;103:198–206.



