






Introducción
El dermatofibrosarcoma protuberans (DFSP) es un tumor fibrohistiocitario de bajo grado que se caracteriza por una elevada tasa de recurrencias locales. Se suele presentar entre los 20 y 50 años de edad, siendo raro en los niños menores de 16 años y todavía más raro los casos congénitos, con sólo 27 casos descritos en la literatura1, 2. Se describe el caso de un niño de 10 años de edad con un dermatofibrosarcoma protuberans presente desde el nacimiento y sin signos de recurrencia en la actualidad.
Caso clínico
Se trata de un niño de 10 años de edad de origen marroquí y sin antecedentes médicos de interés, que consultaba por una tumoración en la pierna izquierda presente desde el nacimiento y que le había ido creciendo progresivamente. En la exploración física se objetivaba una placa indurada de 9 × 7,5 cm de diámetro situada en la cara latero-interna de la pierna izquierda (fig. 1). La placa presentaba dos áreas periféricas atróficas que dejaban ver la red vascular subyacente. En el centro se observaban dos nódulos tumorales de 1 cm de diámetro de aspecto pardo-amarillento y otro violáceo de mayor tamaño (fig. 2). A la palpación la lesión era firme y estaba unida al tejido subcutáneo. Una radiografía ósea de la tibia y el peroné y una ecografía de partes blandas fueron normales. Se practicó una biopsia cutánea que mostró una epidermis atrófica y un infiltrado muy celular que se extendía en profundidad desde la dermis papilar (fig. 3). El infiltrado estaba constituido por células fusiformes de núcleo alargado que se disponían en fascículos cortos con un patrón estoriforme (fig. 4). En la inmunohistoquímica las células del tumor fueron positivas con el anticuerpo para CD34 y la tenascina (fig. 5). Esta histología con esta inmunohistoquímica correspondían a un dermatofibrosarcoma protuberans, que en este caso al estar presente desde el nacimiento tenía además la peculiaridad de ser congénito. Tras el diagnóstico, al niño se le realizó una exéresis amplia del tumor que por motivos personales se realizó en otro centro, y hasta el momento no tenemos constancia de recidiva.
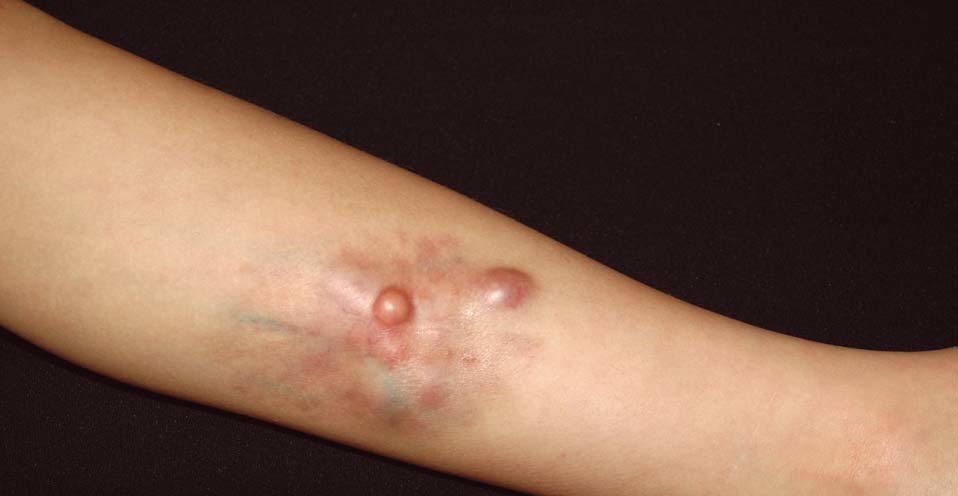
Figura 1. Placa indurada localizada en la cara latero-interna de la pierna izquierda.
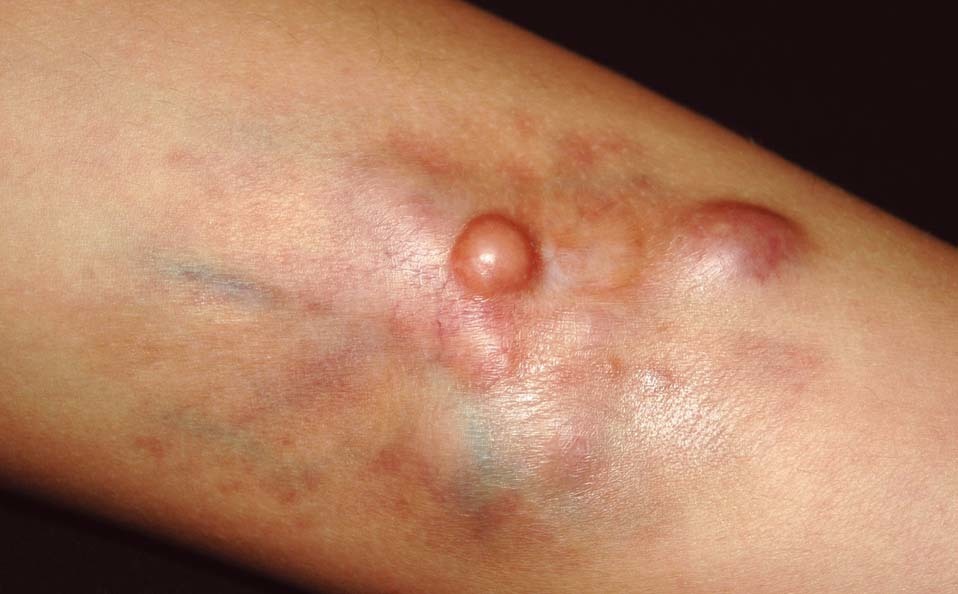
Figura 2. La placa presenta un área periférica atrófica que deja ver la red vascular subyacente y en el centro se observan dos nódulos pardo-amarillentos de 1 cm de diámetro y otro violáceo de mayor tamaño.
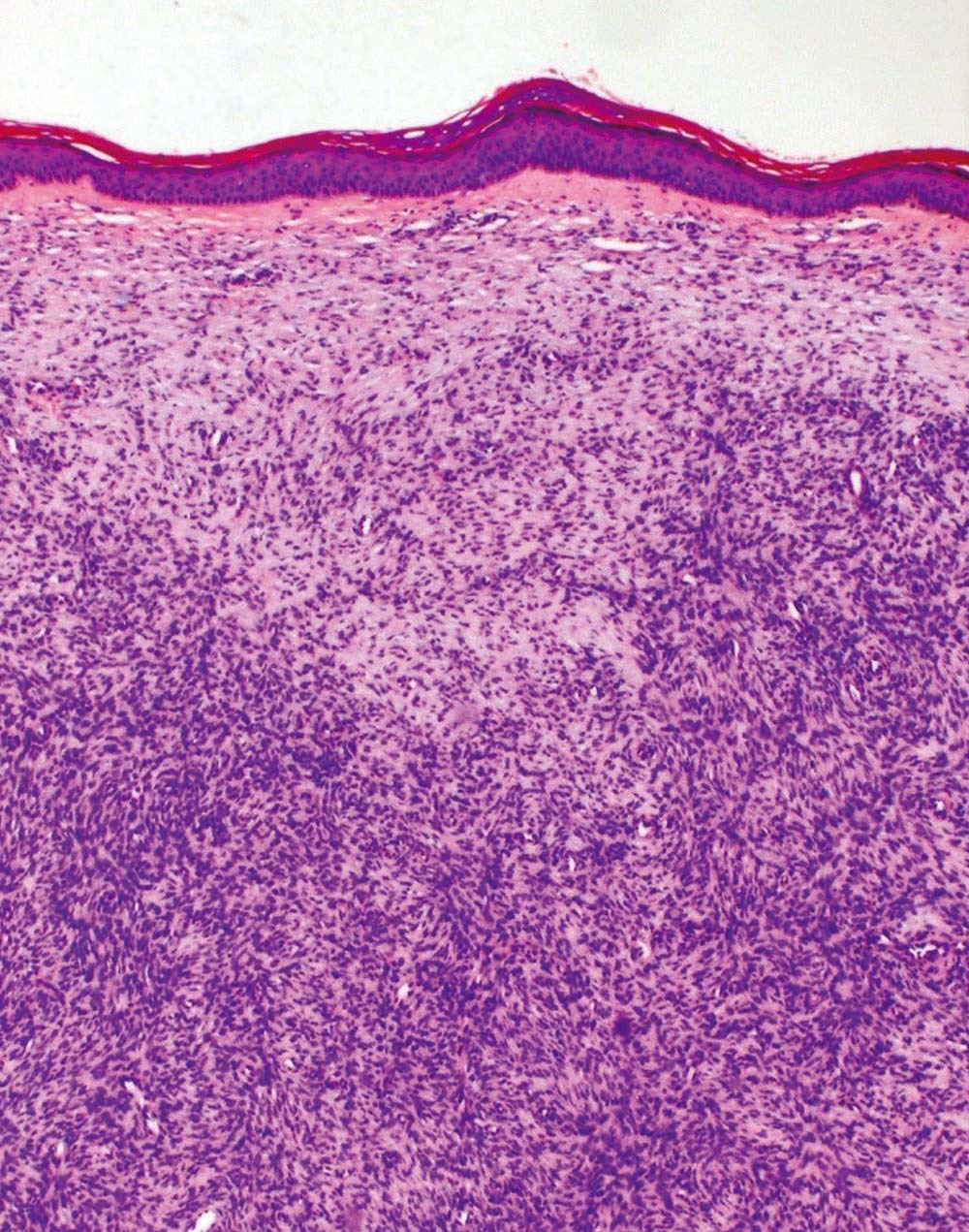
Figura 3. En la histología se observa un infiltrado de células fusiformes que se extiende en profundidad desde la dermis papilar. (Hematoxilina-eosina, ×100.)
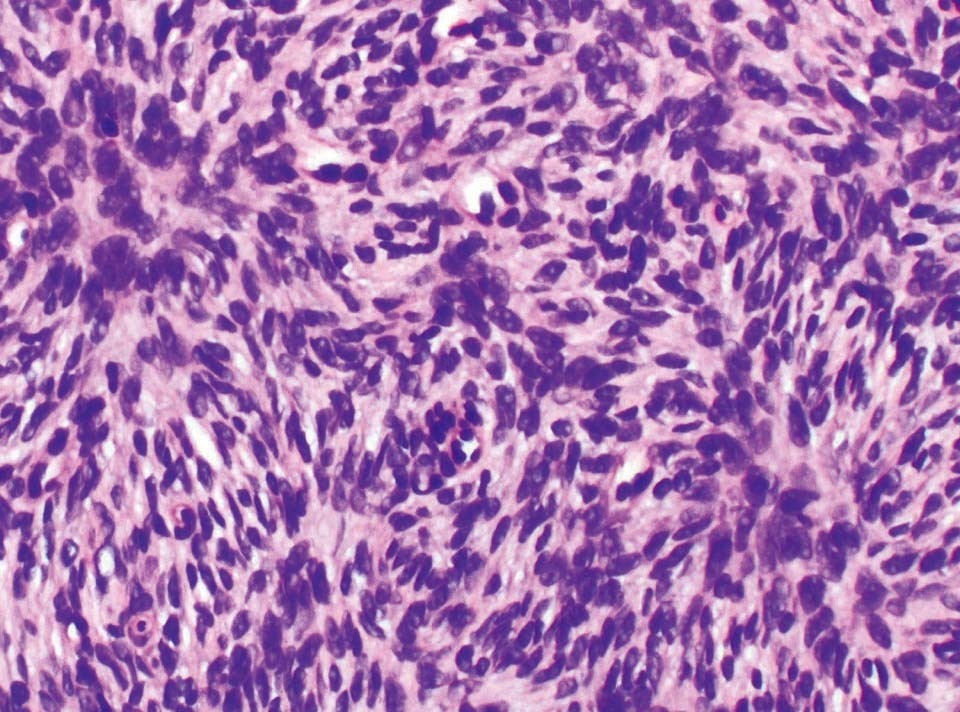
Figura 4. Las células fusiformes del infiltrado se disponen en fascículos cortos adoptando un patrón arremolinado. (Hematoxilina-eosina, ×200.)
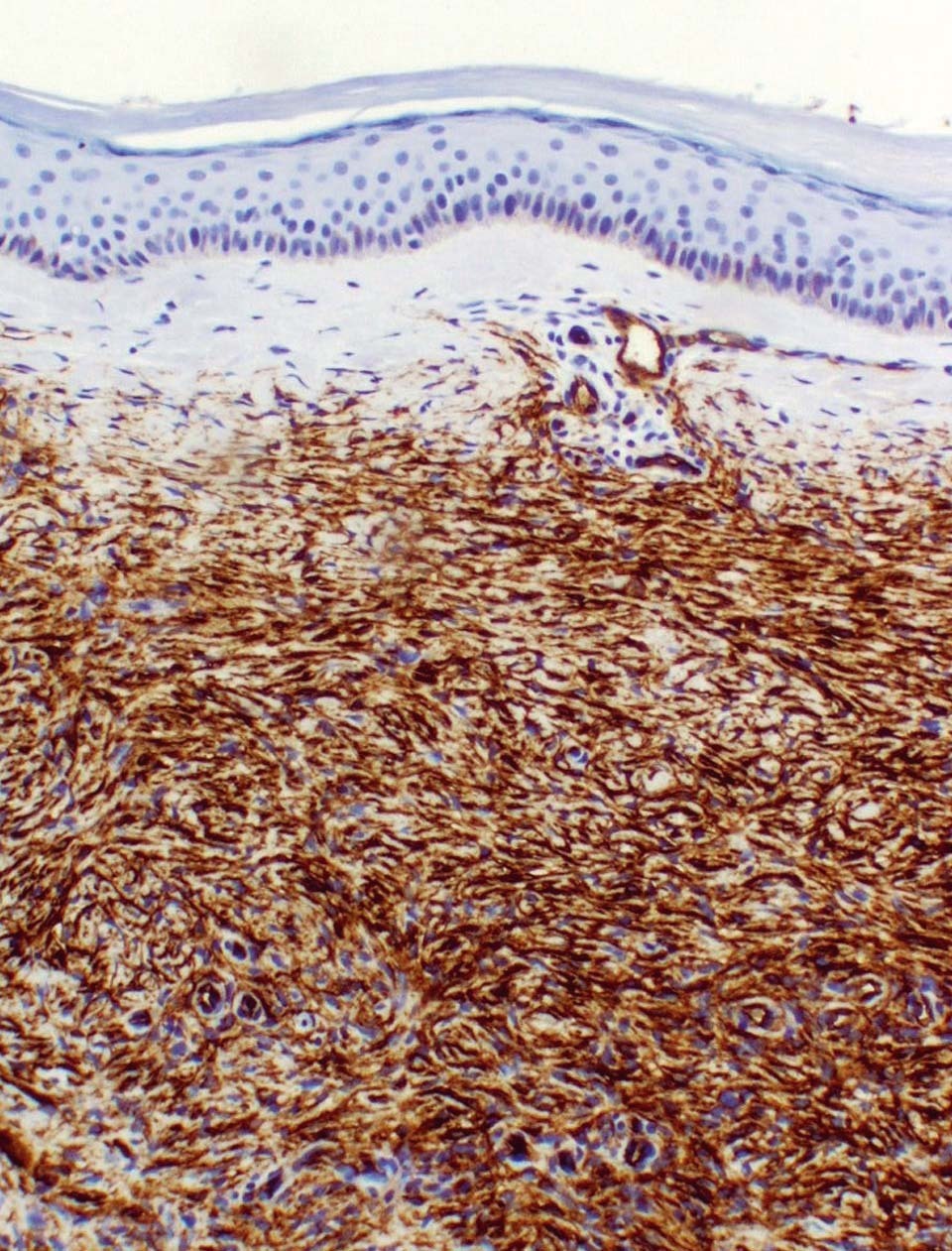
Figura 5. En la inmunohistoquímica las células del tumor son positivas con el anticuerpo para CD34. (Inmunohistoquímica para el marcador CD34, ×100.)
Discusión
El DFSP es un tumor fibrohistiocitario de bajo grado. Fue inicialmente descrito en 1924 por Darier y Ferrand con el nombre de «dermatofibroma progresivo y recurrente» y un año más tarde Hoffman describió tres nuevos casos y propuso el término de «dermatofibrosarcoma protuberans». Es un tumor poco frecuente, con una incidencia del 0,1 % de los tumores malignos3. Se suele presentar entre los 20 y 50 años de edad, siendo raro en los niños menores de 16 años (el 6 % de los DFSP) y todavía más raro los casos congénitos con sólo 27 casos descritos en la literatura1,2. En general hay una mayor incidencia en la raza caucásica. Es ligeramente más frecuente en los varones, aunque en los casos congénitos predomina en el sexo femenino. Las localizaciones más frecuentes son el tronco y la parte proximal de extremidades y es raro en la cabeza y en el cuello. Hay una gran variabilidad clínica en la presentación del DFSP2,4,5. Habitualmente suele presentarse como una mácula o una placa de color eritemato-azulada o violácea de aspecto vascular. Tiene un lento crecimiento, durante años, en el que aparecen pequeños nódulos en la superficie de la placa, con una lenta progresión a un estado nodular o protuberante. Es una placa indurada y asintomática, aunque en ocasiones puede ulcerarse y ser dolorosa. En algunos casos, persiste como una placa no protuberante, recibiendo entonces la denominación de DFSP atrófico. En estos casos, suele presentarse como una lesión violácea, deprimida y atrófica semejante a una cicatriz, sin ningún nódulo, que con frecuencia se confunde clínicamente con un carcinoma basocelular esclerosante, una anetodermia, una morfea o una cicatriz. El DFSP atrófico es especialmente frecuente en los casos congénitos y en la infancia6.
El diagnóstico del DFSP es histológico7, 8. Se trata de un tumor de células fusiformes de núcleo alargado que se disponen en fascículos cortos adoptando un patrón estoriforme o arremolinado característico. Las células muestran una atipia celular mínima y pocas mitosis. Tiene un patrón de crecimiento infiltrativo y generalmente se extiende hasta la dermis profunda y el tejido celular subcutáneo. El tumor de Bednar es una variante histológica del DFSP, que tiene células dendríticas que contienen melanina entre las células fusiformes9. Otro subtipo es el mixoide, con áreas de mucina intersticial entre las células fusiformes10. En la inmunohistoquímica es muy importante destacar la positividad de las células del tumor con el anticuerpo para CD34. El CD34 es un antígeno de superficie que se expresa en las células madre hematopoyéticas, en el endotelio y en la piel (alrededor de las estructuras foliculares, glándulas sebáceas y sudoríparas). Este antígeno puede estar presente también en neoplasias como ocurre en el DFSP y es de gran ayuda para diferenciarlo de otros tumores fibrohistiocitarios. La tenascina es un marcador estromal de tejido conectivo que suele ser positivo en las células del DFSP y es característico que la positividad se extienda hacia el tejido celular subcutáneo. Vimentina también es positiva y el resto de marcadores (factor XIIIa, S-100, actina y desmina) son negativos. Citogenéticamente, el DFSP se caracteriza por presentar la translocación recíproca t(17;22)(q22;q13) que condiciona en la fusión del gen del colágeno tipo 1 (COL1A1), en el cromosoma 17q, con el gen de la cadena beta del factor de crecimiento derivado de las plaquetas (PDGFB), en el cromosoma 22q1, 3 11.
El diagnóstico clínico del DFSP en la infancia o en los jóvenes puede ser dificultoso a causa de que en los estadios tempranos el tumor frecuentemente se parece a una malformación vascular. El resto de diagnósticos diferenciales deben hacerse con los tumores fibrohistiocitarios, como dermatofibroma, leiomioma, neurofibroma, dermatomiofibroma, y en la infancia también con la miofibromatosis infantil y el hamartoma fibroso de la infancia1,2,7. El dermatofibroma es una lesión más nodular, delimitada a la dermis y si invade el tejido celular subcutáneo lo hace por los septos interlobulillares, y en la inmunohistoquímica, al contrario de lo que ocurre en el DFSP, es CD34 negativo y factor XIIIa positivo. El leiomioma está constituido por células fusiformes que poseen núcleos con morfología de cigarro puro y se disponen paralelas a la epidermis. Si fuera necesario recurrir a la inmunohistoquímica, el leiomioma es positivo para desmina y caldesmón, ambos negativos en el DFSP. El neurofibroma presenta unos núcleos más pequeños y ondulados y es positivo para S-100, anticuerpo constantemente negativo en el DFSP. El dermatomiofibroma es un tumor raro que se presenta como una placa asintomática de crecimiento lento. Es frecuente en el cuello y en los hombros de adultos jóvenes, con predilección por las mujeres. En la histología el tumor tiene aspecto de placa, bien circunscrita, compuesta por fascículos de células monomorfas fusiformes, orientadas paralelamente a la epidermis. Las células presentan citoplasma eosinófilo y núcleo elongado vesiculoso con 1 o 2 nucléolos y expresan vimentina y actina de músculo liso. La miofibromatosis infantil se caracteriza por la aparición de lesiones nodulares o tumorales, firmes y elásticas, de localización preferente en tronco, hombros y muslos. Existen formas solitarias, tanto a nivel cutáneo como óseo o visceral. En la histología las células fusiformes tienen un citoplasma eosinófilo y núcleos vesiculosos que se disponen en fascículos cortos entrelazados. Los agregados celulares están separados por finas bandas de colágeno y a menudo, en el centro de la tumoración se observan espacios vasculares. Desde el punto de vista inmunohistoquímico, las células son positivas para vimentina y actina. El hamartoma fibroso de la infancia es una lesión prácticamente exclusiva de la época prepuberal, siendo muy característico su patrón organoide constituido por trabéculas de tejido fibrocolágeno, células primitivas mesenquimales y tejido adiposo maduro, y es fácil diferenciarlo del DFSP con la simple microscopía óptica.
El tratamiento del DFSP consiste en la exéresis amplia del tumor con 2-3 cm de margen incluyendo la fascia superficial. La radioterapia se reserva para las escisiones incompletas o para tumores de gran tamaño como terapia coadyuvante. El DFSP es un tumor que se caracteriza por una marcada invasión local y una elevada tasa de recurrencias locales (entre el 20-50 %), sobre todo dentro de los tres primeros años después de la intervención quirúrgica. La microcirugía de Mohs ha disminuido la tasa de recurrencias a menos del 2 % y se considera el tratamiento de elección1,9. La resonancia magnética, según estudios recientes, puede ser de gran ayuda como instrumento preoperatorio para delimitar la extensión y el tamaño del tumor y planificar así la cirugía y prevenir las recurrencias locales1. También puede ayudar en el seguimiento posoperatorio para detectar y monitorizar las recidivas del tumor. Se recomiendan controles clínicos de los pacientes cada 3-6 meses en los tres primeros años después de la intervención y posteriormente una vez al año. Las metástasis del DFSP son extremadamente raras. Han sido reportadas en el 1- 6 % de los pacientes y son un signo de mal pronóstico1,7. El pulmón es el sitio más común de las metástasis por vía hematógena, seguido de las adenopatías regionales, el cerebro, el hueso y el corazón.
Como conclusiones, debemos destacar que ante un niño que se presente con placas o nódulos cutáneos, incluso congénitos, que no tengan un diagnóstico clínico claro, es recomendable hacer una biopsia diagnóstica. Además, dado el potencial local agresivo y la alta tasa de recurrencias del DFSP, es importante un diagnóstico temprano para facilitar una escisión adecuada.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Cristina Muniesa.
Servicio de Dermatología.
Hospital Mútua de Terrassa.
Pl. Doctor Robert, 5.
08221 Terrassa. Barcelona. España.
cristinamuniesa@hotmail.com
Aceptado el 8 de noviembre de 2006.



