







La epidermólisis bullosa (EB) es un grupo heterogéneo de trastornos hereditarios caracterizado por un aumento de la fragilidad mucocutánea. El objetivo del presente estudio es describir las características clínicas y epidemiológicas de los pacientes con EB atendidos en el Hospital Universitario La Paz, centro de referencia nacional para EB hereditaria.
Material y métodoEstudio observacional, retrospectivo y unicéntrico. Se incluyeron todos los pacientes con diagnóstico clínico y molecular de EB atendidos en el Servicio de Dermatología del Hospital Universitario La Paz desde el 1 de enero de 2000 hasta el 28 de febrero de 2021.
ResultadosSe registraron 214 pacientes, con una edad mediana de 17 años (RIQ: 8-32); el 54,2% fueron mujeres. Las formas clínicas correspondieron a EB distrófica con 135 (63,1%) casos, EB simple con 67 (31,3%) casos, EB juntural con ocho (3,7%), EB Kindler con tres (1,4%) casos y EB adquirida con un (0,5%) caso. El 35,5% de los pacientes procedían de Madrid. Las complicaciones clínicas más frecuentes en nuestra serie fueron el prurito (63,1%), las infecciones locales (56,5%) y el dolor (54,7%). Las complicaciones más graves fueron las cardíacas (5,6%) y la aparición de CCE (10,3%). Fallecieron 22 pacientes (10,3%).
ConclusionesLa forma clínica predominante fue la EBDR. Las complicaciones más prevalentes fueron el prurito, el dolor y las infecciones, y las más graves, la miocardiopatía y el CCE. Es un estudio pionero realizado en nuestro país que permitirá implementar estrategias para mejorar la situación sociosanitaria de los pacientes con EB.
Epidermolysis bullosa (EB) is a heterogeneous group of inherited disorders characterized by a high degree of mucocutaneous fragility. This study aimed to describe the clinical and epidemiologic characteristics of patients with EB treated in Hospital Universitario La Paz, a national referral center for inherited EB.
Material and methodsObservational, retrospective, single-center study. We included all cases with a clinical and molecular diagnosis of EB managed in the hospital's dermatology department from January 2, 2000, to February 28, 2021.
ResultsA total of 214 cases were studied. The median (interquartile range) age was 17 (8–32) years; 54.2% were women. One hundred thirty-five (63.1%) patients had dystrophic EB, 67 (31.3%) had EB simplex, 8 (3.7%) had junctional EB, and 3 (1.4%) had Kindler syndrome. One (0.5%) had EB acquisita. Over a third (35.5%) of the patients resided in Madrid. The most common clinical complications were pruritus (63.1%), local infections (56.5%), and pain (54.7%). The most serious ones were cardiomyopathy (in 5.6%) and squamous cell carcinoma (10.3%). Twenty-two patients (10.3%) died.
ConclusionsDystrophic EB was the most prevalent clinical form. The most prevalent complications were pruritus, pain, and infections. The most serious ones were cardiomyopathy and squamous cell carcinoma. This study is the first in Spain that explores strategies for improving the health status and quality of life of patients with EB.
La epidermólisis bullosa (EB) es un grupo heterogéneo de trastornos hereditarios caracterizado por un aumento de la fragilidad mucocutánea, con aparición de ampollas de forma espontánea o ante traumatismos mecánicos mínimos1. Se considera una enfermedad rara, con una prevalencia estimada en Europa de seis por cada millón de habitantes2.
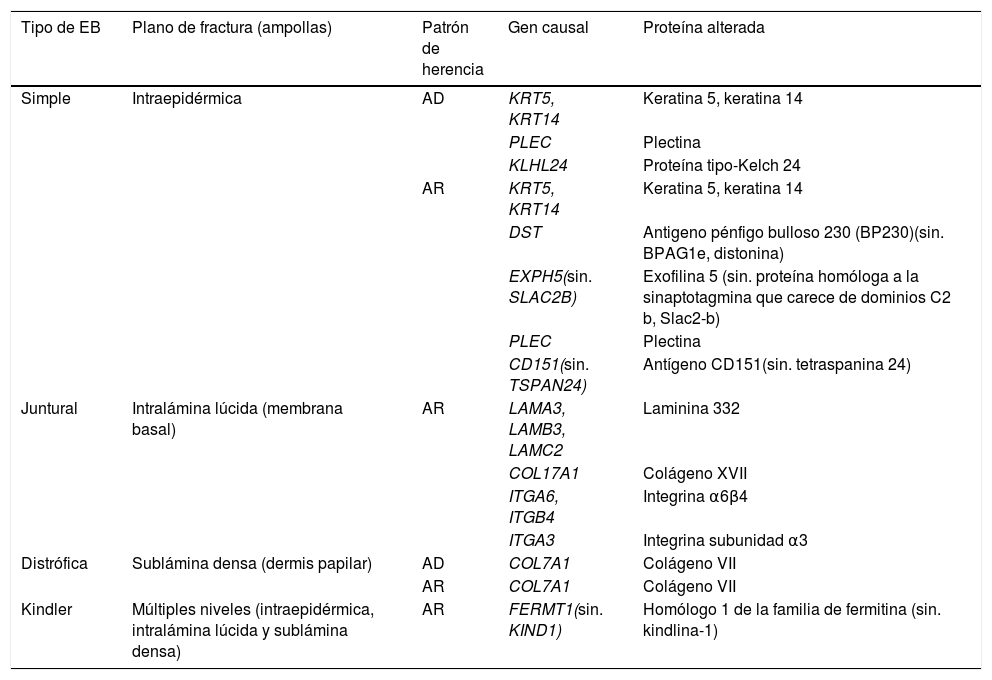
La EB está causada por mutaciones en genes que codifican proteínas responsables de la integridad y la estabilidad mecánica del tegumento3,4. Hasta la fecha, se han documentado más de 1.000 mutaciones en 21 genes estructurales que resultan en una adhesión defectuosa de la piel y la consecuente fragilidad cutánea, dando lugar a diversas formas de EB con afectación cutánea y extracutánea1,5,6. Debido al gran número de proteínas involucradas en esta enfermedad, el diagnóstico de la EB es complejo y su clasificación ha estado sujeta a revisiones internacionales periódicas. En la última clasificación, se han descrito más de 30 subtipos de EB, que se agrupan en dos categorías distintas5,6. La primera categoría corresponde a las formas clásicas de EB, definidas según el nivel de formación de la ampolla dentro de la piel, e incluye a la EB simple (EBS), EB juntural (EBJ), EB distrófica (EBD) y EB Kindler (EBK) (tabla 1; fig. 1); la segunda categoría corresponde a otros trastornos con tendencia a la formación de ampollas, pero donde la patología primaria y las manifestaciones clínicas son extracutáneas (tabla 2). Las características específicas de los subtipos clásicos de EB se resumen en la tabla 3 (material suplementario).
Características moleculares de los cuatro tipos de EB clásicos
| Tipo de EB | Plano de fractura (ampollas) | Patrón de herencia | Gen causal | Proteína alterada |
|---|---|---|---|---|
| Simple | Intraepidérmica | AD | KRT5, KRT14 | Keratina 5, keratina 14 |
| PLEC | Plectina | |||
| KLHL24 | Proteína tipo-Kelch 24 | |||
| AR | KRT5, KRT14 | Keratina 5, keratina 14 | ||
| DST | Antigeno pénfigo bulloso 230 (BP230)(sin. BPAG1e, distonina) | |||
| EXPH5(sin. SLAC2B) | Exofilina 5 (sin. proteína homóloga a la sinaptotagmina que carece de dominios C2 b, Slac2-b) | |||
| PLEC | Plectina | |||
| CD151(sin. TSPAN24) | Antígeno CD151(sin. tetraspanina 24) | |||
| Juntural | Intralámina lúcida (membrana basal) | AR | LAMA3, LAMB3, LAMC2 | Laminina 332 |
| COL17A1 | Colágeno XVII | |||
| ITGA6, ITGB4 | Integrina α6β4 | |||
| ITGA3 | Integrina subunidad α3 | |||
| Distrófica | Sublámina densa (dermis papilar) | AD | COL7A1 | Colágeno VII |
| AR | COL7A1 | Colágeno VII | ||
| Kindler | Múltiples niveles (intraepidérmica, intralámina lúcida y sublámina densa) | AR | FERMT1(sin. KIND1) | Homólogo 1 de la familia de fermitina (sin. kindlina-1) |
EB: Epidermólisis bullosa; AD: autosómica dominante; AR: autosómica recesiva
, EBJ (Fig. 1B), EBD (Fig. 1C) y EBK (Fig. 1D).")
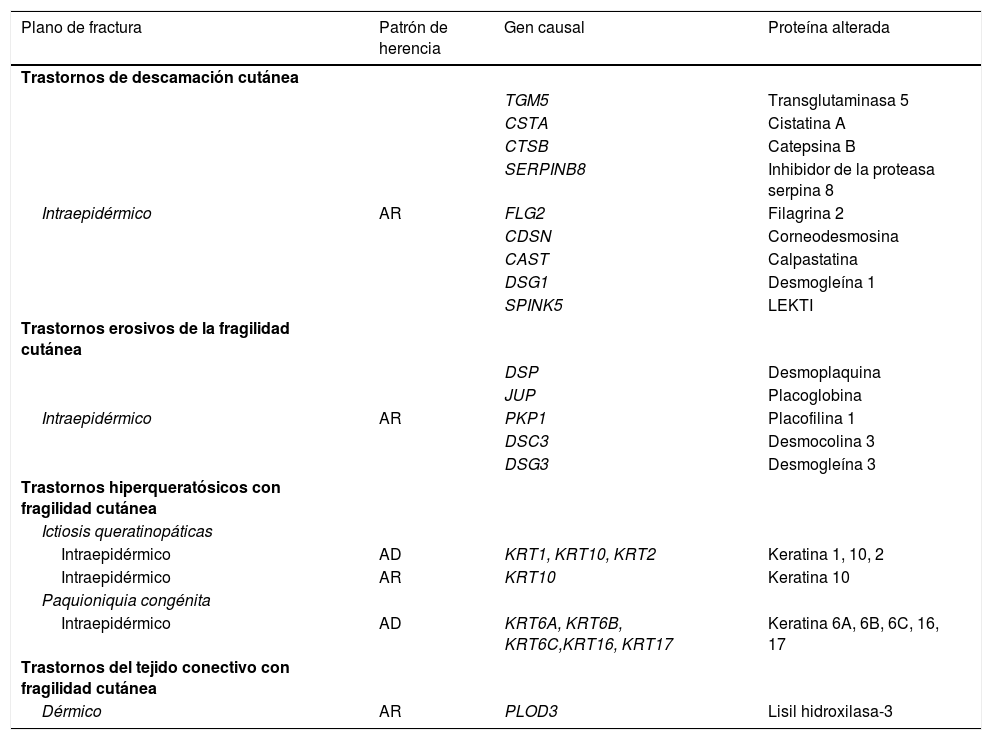
Características moleculares de trastornos genéticos con fragilidad cutánea menor
| Plano de fractura | Patrón de herencia | Gen causal | Proteína alterada |
|---|---|---|---|
| Trastornos de descamación cutánea | |||
| TGM5 | Transglutaminasa 5 | ||
| CSTA | Cistatina A | ||
| CTSB | Catepsina B | ||
| SERPINB8 | Inhibidor de la proteasa serpina 8 | ||
| Intraepidérmico | AR | FLG2 | Filagrina 2 |
| CDSN | Corneodesmosina | ||
| CAST | Calpastatina | ||
| DSG1 | Desmogleína 1 | ||
| SPINK5 | LEKTI | ||
| Trastornos erosivos de la fragilidad cutánea | |||
| DSP | Desmoplaquina | ||
| JUP | Placoglobina | ||
| Intraepidérmico | AR | PKP1 | Placofilina 1 |
| DSC3 | Desmocolina 3 | ||
| DSG3 | Desmogleína 3 | ||
| Trastornos hiperqueratósicos con fragilidad cutánea | |||
| Ictiosis queratinopáticas | |||
| Intraepidérmico | AD | KRT1, KRT10, KRT2 | Keratina 1, 10, 2 |
| Intraepidérmico | AR | KRT10 | Keratina 10 |
| Paquioniquia congénita | |||
| Intraepidérmico | AD | KRT6A, KRT6B, KRT6C,KRT16, KRT17 | Keratina 6A, 6B, 6C, 16, 17 |
| Trastornos del tejido conectivo con fragilidad cutánea | |||
| Dérmico | AR | PLOD3 | Lisil hidroxilasa-3 |
AD: autosómica dominante; AR: autosómica recesiva.
Características clínicas de los tipos clásicos de EB (material suplementario)
| Subtipo de EB(denominaciones previas) | Fenotipo | Patrón de herencia | Gen causal |
|---|---|---|---|
| EB simple | |||
| EB simple, localizada(EBS Weber-Cockayne) | Ampollas palmo-plantares desde el nacimiento o la primera infancia, generalmente estacionales, con desarrollo posterior de queratodermia en las zonas afectadas | AD | KRT5 o KRT14 |
| EB simple grave(EBS generalizada grave, EBS Dowling-Meara) | Ampollas generalizadas tempranas y zonas de aplasia, en ocasiones desde el nacimiento o días después; puede ser mortal en el primer año de vida; clásicamente, surgen ampollas «herpetiformes» tensas agrupadas tras un traumatismo mínimo o de forma espontánea; desarrollo de queratodermia palmo-plantar confluente; distrofia ungueal común | AD | KRT5 o KRT14 |
| EB simple intermedia(EBS generalizada intermedia, EBS Köbner) | Ampollas generalizadas, aunque menos graves que las de EB simple grave | AD | KRT5 o KRT14 |
| EB simple con pigmentación moteada | Ampollas generalizadas desde el nacimiento de gravedad intermedia; pigmentación moteada o reticulada típicamente en el cuello, tronco superior y piel acral; queratodermia palmo-plantar y distrofia ungueal años después | AD | KRT5>KRT14 |
| EB simple circinada migratoria | Vesículas desde el nacimiento, que adquieren con el tiempo un patrón migratorio circinado sobre un fondo eritematoso, dando lugar después a hiperpigmentación postinflamatoria; posible distrofia ungueal | AD | KRT5 |
| EB simple intermedia con cardiomiopatía | Erosiones marcadas en las extremidades al nacer, que curan con despigmentación y cicatrices atróficas; puede aparecer queratodermia, engrosamiento de las uñas y onicogrifosis; ocasionalmente se ha notificado alopecia difusa; la cardiomiopatía dilatada se desarrolla más tarde en la edad adulta joven | AD | KLHL24 |
| EB simple intermedia con mutaciones en PLEC | La enfermedad autosómica dominante es leve y presenta principalmente ampollas acrales; la autosómica recesiva tiene una presentación intermedia | AD o AR | PLEC |
| EB simple intermedia con distrofia muscular | Ampollas generalizadas con miopatía de inicio variable que incluye distrofia muscular y una posible cardiomiopatía; queratodermia plantar focal y distrofia ungueal; la afectación de las mucosas es frecuente; puede aparecer estenosis del tracto respiratorio superior | AR | PLEC |
| EB simple con atresia de píloro | Ausencia congénita generalizada de piel en todo su espesor o ampollas generalizadas con atresia pilórica; mortalidad temprana a los pocos meses de vida | AR | PLEC |
| EB simple autosómica recesiva KRT5 o KRT14 | Ampollas al nacimiento generalizadas y graves en la mayoría de los casos. No existe una mejoría de la fragilidad cutánea con la edad. La cicatrización de las lesiones da lugar a una hiperpigmentación postinflamatoria; la ausencia de queratina 5 da lugar a ampollas y erosiones generalizadas y a una letalidad temprana | AR | KRT5 o KRT14 |
| EB simple localizada o intermedia con deficiencia de BP230 | Ampollas de inicio temprano, generalmente con predominio acral; queratodermia plantar | AR | DST |
| EB simple localizada o intermedia con deficiencia de exofilina 5 | Ampollas intermitentes generalizadas y fragilidad de la piel; puede ser evidente una leve pigmentación moteada | AR | EXPH5 |
| EB simple localizada con nefropatía (deficiencia de CD151) | Ampollas al nacimiento de predominio pretibial; puede observarse poiquilodermia; alopecia precoz; la afectación extracutánea se manifiesta en forma de membranas esofágicas y nefropatía | AR | CD151 |
| EB juntural | |||
| EB juntural grave(EBJ generalizada grave, EBJ de Herlitz) | Las ampollas pueden ser leves al nacer y estar localizadas en la zona periungueal, nalgas y codos; con los días, las heridas pueden volverse crónicas con un lecho de tejido de granulación friable, que suele afectar a la cara, orejas y parte distal de los dedos; la alopecia es común; los defectos del esmalte dental son habituales; el llanto ronco es a menudo una característica; suele ser mortal en los primeros dos años de vida | AR | LAMA3, LAMB3 y LAMC2 |
| EB juntural intermedia(EBJ generalizada intermedia, EBJ no de Herlitz) | Las ampollas son generalizadas, pero menos severas, en la EBJ intermedia, normalmente sin tendencia a desarrollar tejido de granulación crónico; riesgo elevado de carcinoma epidermoide en la edad adulta | AR | LAMA3, LAMB3, LAMC2 y COL17A |
| EB juntural con atresia de píloro | Se observan extensas áreas de pérdida de piel al nacer con una gran fragilidad cutánea; atresia pilórica de aparición temprana, que supone una causa frecuente de mortalidad temprana, a los pocos días o semanas de nacer; también puede presentarse atresia duodenal duodenal y anal; las variantes más leves variantes no letales suelen presentar afectación genitourinaria | AR | ITGA6 y ITGB4 |
| EB juntural localizada | Fragilidad cutánea limitada, a menudo acral; defectos ungueales y dentales variables; pelo normal | AR | LAMA3, LAMB3, LAMC2, COL17A1, ITGB4 y ITGA3 |
| EB juntural inversa | Ampollas desde el nacimiento en zonas de flexión; anomalías dentales y pérdida de uñas | AR | LAMA3, LAMB3 y LAMC2 |
| EB juntural de inicio tardío | Inicio en la infancia, a menudo con fragilidad acral; la fragilidad de la piel es progresiva y puede observarse la pérdida de dermatoglifos debido a la cicatrización; defectos variables del esmalte dental y de las uñas | AR | COL17A1 |
| EB juntural síndrome laringo-ónico-cutáneo (LOC) | Fragilidad cutánea desde el nacimiento con tejido de granulación exuberante, sobre todo en cara y cuello; distrofia y pérdida ungueal con tejido de granulación en los lechos ungueales; la granulación laríngea puede llevar a un compromiso respiratorio y a la muerte; granulación conjuntival y de los párpados con el consiguiente simbléfaron, cicatrización y pérdida visual | AR | LAMA3 |
| EB juntural con enfermedad pulmonar intersticial y síndrome nefrótico | Grado variable de afectación cutánea; es frecuente la mortalidad en la primera infancia; es posible la distrofia de las uñas; puede haber pérdida de cabello | AR | IGTA3 |
| EB distrófica | |||
| EBDD intermedia(EBDD generalizada) | Fragilidad cutánea generalizada, cicatrices y milia que se presentan desde el nacimiento o en la primera infancia, con prominencia sobre las zonas acrales, los codos y las rodillas; la afectación de las mucosas puede provocar microstomía, anquiloglosia y estenosis esofágica, aunque con menor frecuencia que en la EBDR grave | AD | COL7A1 |
| EBDD localizada(EBDD solo uñas, EBDD pretibial y acral) | Predominan las ampollas acrales, las cicatrices y milia desde el nacimiento o en la primera infancia; ocasionalmente se presenta solo en las uñas, con distrofia progresiva y eventual pérdida de las mismas; raramente, las características cutáneas pueden predominar solo en la piel pretibial (y puede presentarse como enfermedad de inicio tardío) | AD | COL7A1 |
| EBDD pruriginosa | Aparición de pápulas violáceas muy pruriginosas, excoriadas o placas lineales y cicatrices con milia, especialmente en la parte inferior de las piernas, los muslos y los brazos; pueden presentarse en la infancia o en la edad adulta; la distrofia ungueal es habitual | AD | COL7A1 |
| EBDD autorresolutiva | Ampollas evidentes al nacimiento o poco después, generalmente en las extremidades donde puede haber aplasia cutis y aparecer cicatrices y milia; resolución espontánea de la fragilidad cutánea en los primeros dos años de vida | AD | COL7A1 |
| EBDR intermedia(EBDR intermedia generalizada, EBDR no Hallopeau-Siemens) | Fenotipo similar al de la EBDD intermedia, aunque de mayor gravedad con contracturas de flexión, fusión digital proximal limitada y queratodermia estriada ocasional en los dedos | AR | COL7A1 |
| EBDR grave(EBDR grave generalizada, EBDR Hallopeau-Siemens) | Ampollas generalizadas desde el nacimiento, con extensas cicatrices y desarrollo de microstomía, anquiloglosia, estenosis esofágica, contracturas en flexión de las extremidades y pseudosindactilia; pérdida frecuente de las uñas al principio del curso de la enfermedad; alto riesgo de aparición de carcinoma epidermoide cutáneo sobre las heridas crónicas | AR | COL7A1 |
| EBDR inversa | Ampollas generalizadas desde el nacimiento, de gravedad intermedia; posteriormente, la fragilidad tiende a manifestarse en las zonas de flexión | AR | COL7A1 |
| EBDR localizada | Fragilidad de la piel y formación de ampollas típicamente en el nacimiento o en el periodo neonatal, limitadas a sitios acrales como las manos y los pies, u ocasionalmente solo a la zona pretibial, donde puede manifestarse como enfermedad de aparición tardía durante la edad adulta; distrofia de las uñas con anoniquia habitual | AR | COL7A1 |
| EBDR pruriginosa | Similar a la EBDD pruriginosa | AR | COL7A1 |
| EBDR autorresolutiva | Similar a la EBDD autorresolutiva | AR | COL7A1 |
| EBD grave | Clínicamente indistinguible de la EBDR severa, con fragilidad mucocutánea grave desde el nacimiento | Heterocigosidad compuesta dominante y recesiva | COL7A1 |
| EB Kindler | |||
| (Síndrome de Kindler) | Ampollas generalizadas y fotosensibilidad variable desde el nacimiento o la primera infancia, con fragilidad de las mucosas; las ampollas dan paso a una poiquilodermia progresiva, inicialmente más marcada en el dorso de las manos y el cuello; pueden aparecer queratodermia palmo-plantar y pérdida de dermatoglifos; también son frecuentes la gingivitis y enfermedades dentales; hay aumento de riesgo de carcinoma epidermoide mucocutáneo, con mal pronóstico | AR | FERMT1 |
EB: epidermólisis bullosa; EBS: epidermólisis bullosa simple; EBJ: epidermólisis bullosa juntural; EBDD: epidermólisis bullosa distrófica dominante; EBDR: epidermólisis bullosa distrófica recesiva; AD: autosómica dominante; AR: autosómica recesiva.
La EB abarca un amplio espectro de fenotipos, que va desde la aparición de ampollas en palmas y plantas, hasta complicaciones extracutáneas que pueden poner en riesgo la vida de los pacientes (fig. 2). Los epitelios con mayor riesgo de complicaciones son el ocular, las vías respiratorias superiores, el esófago y el tracto genitourinario7. En determinados subtipos de EB pueden surgir complicaciones graves en el sistema músculo-esquelético, el corazón, la médula ósea y la cavidad oral, que contribuyen a un aumento de la morbimortalidad. Además, pueden desarrollarse carcinomas de células escamosas (CCE) potencialmente mortales a partir de la segunda década de vida, principalmente en pacientes con EBD recesiva (EBDR)8.
; pseudosindactilia en manos y pies (Fig. 2B); carcinoma epidermoide en rodilla de paciente con EBDR (Fig. 2C); úlcera corneal epitelial fluoresceína positiva (Fig. 2D).")
A pesar de los enormes progresos realizados en la comprensión de las bases moleculares de las distintas formas de EB, no existe aún una cura para esta enfermedad, y solo es posible hacer un tratamiento preventivo y sintomático de las lesiones cutáneas y de las posibles complicaciones sistémicas.
En el año 2017 fueron designados centros de referencia (CSUR) para EB hereditaria en España, el Hospital Universitario La Paz en Madrid y los hospitales Sant Joan de Déu y Clínic en Cataluña. El objetivo del presente estudio es describir las características clínicas y epidemiológicas de los pacientes con EB atendidos en el Hospital Universitario La Paz en los últimos 20 años.
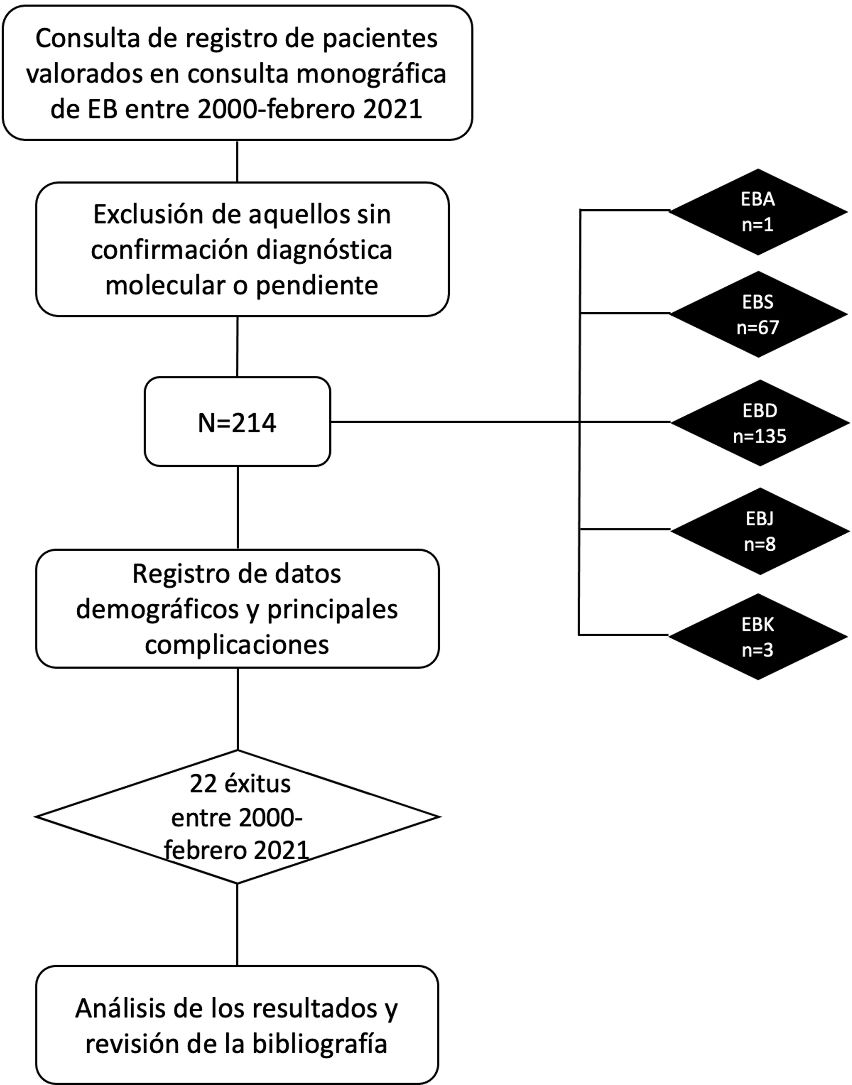
MétodosSe realizó un estudio observacional, retrospectivo y unicéntrico en el Hospital Universitario La Paz. Se incluyeron en el estudio todos los pacientes con diagnóstico clínico y molecular de EB que recibieron atención médica ambulatoria en la Unidad de EB del Servicio de Dermatología del Hospital Universitario La Paz desde el 1 de enero de 2000 hasta el 28 de febrero de 2021.
Se recogieron los datos demográficos de los pacientes, el estudio molecular, el subtipo de EB y las principales complicaciones mucocutáneas y extracutáneas observadas a lo largo del seguimiento de los pacientes. En el análisis estadístico, se utilizaron proporciones y frecuencias absolutas para la descripción de las principales variables cualitativas. Para la estimación de la función de supervivencia, se utilizó el estimador no paramétrico de Kaplan-Meier. Para comparar las curvas, se aplicó la prueba log-rank. El objetivo de este análisis era estudiar la supervivencia global respecto de la mortalidad de los pacientes en cada subtipo de EB.
Este estudio fue autorizado por el Comité Ético del Hospital Universitario La Paz, considerando que el proyecto se limitaba al uso secundario de información previamente obtenida en el curso de la asistencia médica, bajo la responsabilidad del mismo equipo investigador.
ResultadosDurante el período mencionado, fueron atendidos 214 pacientes con EB en el Hospital Universitario La Paz, con una edad mediana de 17 años (rango intercuartílico [RIQ]: 8-32); el 54,2% fueron mujeres. Las formas clínicas correspondieron a EBD con 135 (63,1%) casos, EBS con 67 (31,3%) casos, EBJ con ocho (3,7%) casos, EBK con tres (1,4%) casos y EBA con un (0,5%) caso. El 35,5% de los pacientes procedían de Madrid. Los datos demográficos completos están recogidos en la tabla 4.
Datos demográficos de los pacientes con EB del Hospital Universitario La Paz entre los años 2000-2021 (n = 214)
| n | 214 |
|---|---|
| Sexo (%) | |
| Mujer | 116 (54,2) |
| Varón | 98 (45,8) |
| Subtipo de EB (%) | |
| Simple | 67 (31,3) |
| Distrófica | 135 (63,1) |
| Juntural | 8 (3,7) |
| Kindler | 3 (1,4) |
| Adquirida | 1 (0,5) |
| Edad (mediana, RIQ) | 17,00 (8,00-32,00) |
| Comunidad autónoma de origen (%) | |
| Madrid | 76 (35,5) |
| Castilla y León | 27 (12,6) |
| Andalucía | 26 (12,1) |
| Castilla-La Mancha | 18 (8,4) |
| Extremadura | 13 (6,1) |
| Comunidad Valenciana | 12 (5,6) |
| Galicia | 11 (5,1) |
| País Vasco | 8 (3,7) |
| Canarias | 7 (3,3) |
| Aragón | 6 (2,8) |
| Cataluña | 2 (0,1) |
| Navarra | 2 (0,1) |
| Otros | 6 (2,8) |
EB: epidermólisis bullosa; RIQ: rango intercuartílico.
En el 76,63% de nuestros pacientes se realizó confirmación diagnóstica mediante estudio genético, siendo la mutación patogénica c.6527insC en el gen COL7A1, la más frecuentemente encontrada en nuestra serie, apareciendo en el 21,5% de todos los casos, y en el 34,1% de los pacientes con EBDR.
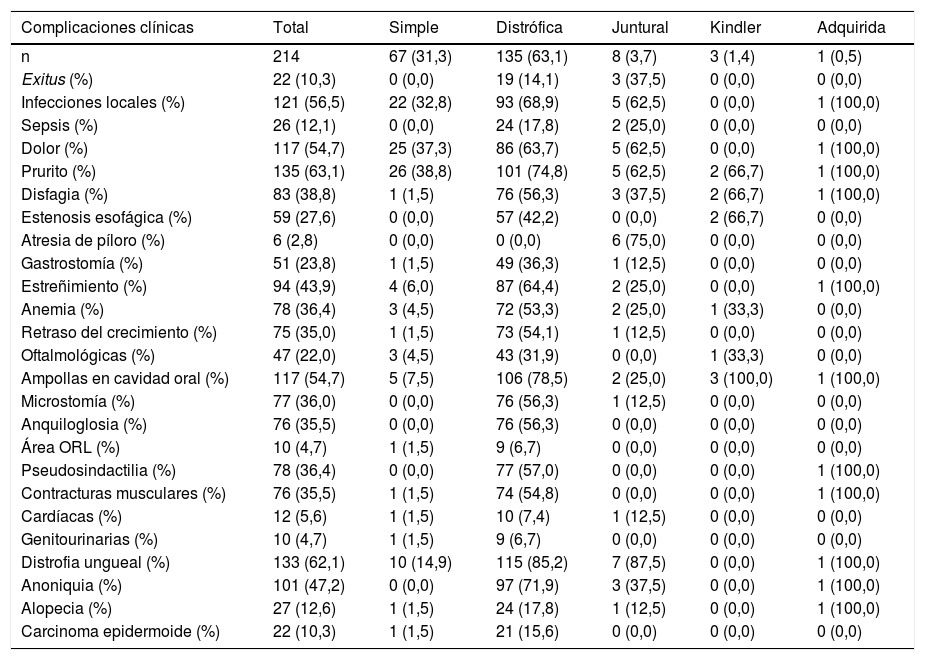
Las principales complicaciones mucocutáneas y extracutáneas observadas en estos pacientes aparecen detalladas en la tabla 5. Las complicaciones más frecuentes en todos los tipos de EB fueron el prurito, que apareció en el 63,1% de los pacientes, y el dolor, encontrado en el 54,7% de los casos. El 56,5% de los individuos presentaron infecciones locales de las heridas, siendo Pseudomonasaeruginosa y Staphylococcusaureus, los microorganismos más frecuentemente implicados (45,22 y 41,44%, respectivamente). Además, 26 pacientes (12,1%) presentaron una sepsis grave, siendo la causa del fallecimiento en dos pacientes con EBJ grave. El 41% de los casos requirió ingreso en la Unidad de Cuidados Intensivos (UCI).
Complicaciones clínicas de EB en los pacientes del Hospital Universitario La Paz entre los años 2000-2021 (n = 214)
| Complicaciones clínicas | Total | Simple | Distrófica | Juntural | Kindler | Adquirida |
|---|---|---|---|---|---|---|
| n | 214 | 67 (31,3) | 135 (63,1) | 8 (3,7) | 3 (1,4) | 1 (0,5) |
| Exitus (%) | 22 (10,3) | 0 (0,0) | 19 (14,1) | 3 (37,5) | 0 (0,0) | 0 (0,0) |
| Infecciones locales (%) | 121 (56,5) | 22 (32,8) | 93 (68,9) | 5 (62,5) | 0 (0,0) | 1 (100,0) |
| Sepsis (%) | 26 (12,1) | 0 (0,0) | 24 (17,8) | 2 (25,0) | 0 (0,0) | 0 (0,0) |
| Dolor (%) | 117 (54,7) | 25 (37,3) | 86 (63,7) | 5 (62,5) | 0 (0,0) | 1 (100,0) |
| Prurito (%) | 135 (63,1) | 26 (38,8) | 101 (74,8) | 5 (62,5) | 2 (66,7) | 1 (100,0) |
| Disfagia (%) | 83 (38,8) | 1 (1,5) | 76 (56,3) | 3 (37,5) | 2 (66,7) | 1 (100,0) |
| Estenosis esofágica (%) | 59 (27,6) | 0 (0,0) | 57 (42,2) | 0 (0,0) | 2 (66,7) | 0 (0,0) |
| Atresia de píloro (%) | 6 (2,8) | 0 (0,0) | 0 (0,0) | 6 (75,0) | 0 (0,0) | 0 (0,0) |
| Gastrostomía (%) | 51 (23,8) | 1 (1,5) | 49 (36,3) | 1 (12,5) | 0 (0,0) | 0 (0,0) |
| Estreñimiento (%) | 94 (43,9) | 4 (6,0) | 87 (64,4) | 2 (25,0) | 0 (0,0) | 1 (100,0) |
| Anemia (%) | 78 (36,4) | 3 (4,5) | 72 (53,3) | 2 (25,0) | 1 (33,3) | 0 (0,0) |
| Retraso del crecimiento (%) | 75 (35,0) | 1 (1,5) | 73 (54,1) | 1 (12,5) | 0 (0,0) | 0 (0,0) |
| Oftalmológicas (%) | 47 (22,0) | 3 (4,5) | 43 (31,9) | 0 (0,0) | 1 (33,3) | 0 (0,0) |
| Ampollas en cavidad oral (%) | 117 (54,7) | 5 (7,5) | 106 (78,5) | 2 (25,0) | 3 (100,0) | 1 (100,0) |
| Microstomía (%) | 77 (36,0) | 0 (0,0) | 76 (56,3) | 1 (12,5) | 0 (0,0) | 0 (0,0) |
| Anquiloglosia (%) | 76 (35,5) | 0 (0,0) | 76 (56,3) | 0 (0,0) | 0 (0,0) | 0 (0,0) |
| Área ORL (%) | 10 (4,7) | 1 (1,5) | 9 (6,7) | 0 (0,0) | 0 (0,0) | 0 (0,0) |
| Pseudosindactilia (%) | 78 (36,4) | 0 (0,0) | 77 (57,0) | 0 (0,0) | 0 (0,0) | 1 (100,0) |
| Contracturas musculares (%) | 76 (35,5) | 1 (1,5) | 74 (54,8) | 0 (0,0) | 0 (0,0) | 1 (100,0) |
| Cardíacas (%) | 12 (5,6) | 1 (1,5) | 10 (7,4) | 1 (12,5) | 0 (0,0) | 0 (0,0) |
| Genitourinarias (%) | 10 (4,7) | 1 (1,5) | 9 (6,7) | 0 (0,0) | 0 (0,0) | 0 (0,0) |
| Distrofia ungueal (%) | 133 (62,1) | 10 (14,9) | 115 (85,2) | 7 (87,5) | 0 (0,0) | 1 (100,0) |
| Anoniquia (%) | 101 (47,2) | 0 (0,0) | 97 (71,9) | 3 (37,5) | 0 (0,0) | 1 (100,0) |
| Alopecia (%) | 27 (12,6) | 1 (1,5) | 24 (17,8) | 1 (12,5) | 0 (0,0) | 1 (100,0) |
| Carcinoma epidermoide (%) | 22 (10,3) | 1 (1,5) | 21 (15,6) | 0 (0,0) | 0 (0,0) | 0 (0,0) |
EB: epidermólisis bullosa.
La afectación de la mucosa oral en forma de anquiloglosia y microstomía apareció en el 36 y 35,5% de los pacientes, respectivamente.
Dentro de las complicaciones gastrointestinales, 83 pacientes (38,8%) presentaron disfagia, todos ellos con formas graves de EBD y EBJ. En un 27,6% de los pacientes se demostró estenosis esofágica, todos con EBDR. El 23,8% de los casos eran portadores de gastrostomía, la mayoría de ellos con formas de EBDR grave. El 43,9% de los pacientes presentó estreñimiento, siendo así el síntoma digestivo más frecuente en todas las formas de EB. Solo seis pacientes (2,8%) presentaron atresia de píloro, todos ellos con EBJ, y 75 (35%) presentaron retraso del crecimiento, la mayoría de ellos con EBDR. Además, 78 pacientes (36,4%) presentaron anemia multifactorial.
Dentro de las complicaciones músculo-esqueléticas, 76 pacientes (35,5%) presentaron contracturas musculares, y 78 (36,4%) pseudosindactilia en manos y/o pies. Las complicaciones oftalmológicas y genitourinarias aparecieron en el 22 y 4,7%, respectivamente. En el caso del compromiso nefrourológico, cinco pacientes tuvieron glomerulonefritis y uno estenosis meatal, todos ellos con EBDR. Las alteraciones cardíacas aparecieron en 12 pacientes (5,6%), todos ellos con formas de EBDR, salvo un caso con EBS y otro con EBJ.
Veintidós pacientes (10,3%) tuvieron CCE, de los cuales, 13 de ellos (6,1%) presentaron solo un tumor y nueve (4,2%) dos o más tumores. Todos los casos correspondieron a formas de EBDR, salvo uno con EBS.
En relación con la mortalidad, 22 pacientes de nuestra cohorte (10,3%) han fallecido, 10 pacientes a causa de una sepsis grave, dos pacientes por insuficiencia renal terminal y 10 pacientes por metástasis de CCE agresivos. Respecto al análisis de supervivencia global respecto de la etiología de EB, se observan diferencias significativas (p = 0,011) en cuanto al exitus, siendo las formas distróficas y junturales las que tienen un mayor número de fallecidos (fig. 3).
 en cuanto al exitus, siendo las formas distrófica y juntural las que tienen un mayor número de fallecidos. Destacan las formas junturales, en las que, a pesar de contar con un número menor de fallecimientos, su tiempo de supervivencia es menor. El grupo de pacientes con EB distrófica es el subtipo con mayor número de fallecimientos, pero con un tiempo de supervivencia global superior al de las formas junturales.")
Análisis de supervivencia por subtipos de EB. Se observan diferencias significativas (p = 0,011) en cuanto al exitus, siendo las formas distrófica y juntural las que tienen un mayor número de fallecidos. Destacan las formas junturales, en las que, a pesar de contar con un número menor de fallecimientos, su tiempo de supervivencia es menor. El grupo de pacientes con EB distrófica es el subtipo con mayor número de fallecimientos, pero con un tiempo de supervivencia global superior al de las formas junturales.
La aparición de ampollas mucocutáneas recurrentes y cicatrices es el principal sello de identidad de los pacientes con EB y lo que va a condicionar la mayoría de las complicaciones.
Los hallazgos obtenidos en nuestra cohorte de pacientes son comparables a otras series de EB realizadas en otros países7,8. El conocimiento detallado de la frecuencia y la repercusión clínica de estas complicaciones, así como de los subtipos de EB con mayor riesgo de desarrollarlas, es fundamental para realizar un diagnóstico y tratamiento precoces.
En el estudio de prevalencia por subtipos de EB, el grupo mayoritario en nuestra muestra fue el de pacientes con EBDR, con 135 (63,1%) casos. Esto podría deberse a que esta forma clínica es la que presenta mayores complicaciones clínicas y, por tanto, es remitida a centros de referencia desde los primeros años de vida. La EBS, que es la forma clínica más frecuente en reportes de otros países2, aparece infrarrepresentada en nuestra muestra. Cabe la posibilidad de un subregistro, debido a que la ausencia de complicaciones graves asociadas con la enfermedad hace que no sean derivados a nuestro centro.
En cuanto al diagnóstico molecular, la mutación patogénica c.6527insC en el gen COL7A1 fue la más frecuentemente encontrada en nuestra cohorte, apareciendo en el 34,1% de los pacientes con EBDR. Este dato es algo inferior al reportado previamente en un estudio poblacional en el que se observó que la mutación patogénica c.6527insC estaba presente en el 46,3% de los alelos de los pacientes españoles con EBDR9. Es, por tanto, una mutación muy frecuente en España, que proviene de un único ancestro común, y que actualmente se analiza como primer cribado para el estudio de pacientes con EBD.
Las manifestaciones que aparecieron con mayor frecuencia en todos los tipos de EB fueron el prurito (63,1%) y el dolor (54,7%). Se ha postulado que las lesiones cutáneas crónicas, junto con la inflamación sistémica que presentan estos pacientes, pueden llegar a producir una disfunción de las fibras sensitivas, ocasionando una neuropatía periférica, que podría ser la base del prurito y del dolor crónico10. La mayor prevalencia de prurito se observó en individuos con EBD, apareciendo en el 74,8% de los casos. Según la literatura, hasta un 93% de los pacientes con EBD presentan este síntoma y, además, es característica su escasa respuesta a antihistamínicos y a otros grupos farmacológicos como antiepilépticos o antidepresivos11,12.
Las complicaciones infecciosas son frecuentes en todas las formas de EB. En nuestra serie, más de la mitad de los pacientes presentaron al menos un episodio de sobreinfección local, siendo más frecuente en las formas de EBD, seguido de EBJ y EBS. P. aeruginosa y S.aureus fueron, al igual que en otros reportes anteriores, los microorganismos más frecuentemente implicados (45,22 y 41,44%, respectivamente)13. La sepsis apareció en el 12,1% de los pacientes, ocasionando la muerte en dos de ellos. Durante la lactancia y la primera infancia esta complicación aparece casi de forma exclusiva en los pacientes con EBJ, con riesgos acumulados en otras series de entre el 11 y el 20% al año de vida14. Mientras que el riesgo es muy bajo en todos los demás subtipos de EB, el riesgo en la EBDR grave aumenta hasta aproximadamente el 8% a los 35 años de edad8.
La afectación de la mucosa oral en forma de anquiloglosia y microstomía apareció en el 36 y 35,5% de los pacientes, respectivamente, y se observaron principalmente en formas de EBDR grave y, en menor medida, en EBJ, al igual que lo ya descrito en la literatura8,15. Estas manifestaciones dan lugar a una disminución de la apertura bucal y dificultad para la masticación y deglución, que contribuyen al deterioro del estado nutricional.
En un 27,6% de los pacientes se demostró estenosis esofágica, todos ellos con EBDR, siendo la disfagia el síntoma principal. Un estudio de 223 pacientes pediátricos con EB reveló una frecuencia del 64,9% de estenosis esofágicas en pacientes con EBDR16, corroborando frecuencias similares en otras series7. Las estenosis esofágicas en los pacientes con EBDR grave se presentan con frecuencia en la infancia, y más de la mitad de ellos presentan síntomas a los 10 años de edad16. El bajo porcentaje encontrado en nuestra serie podría deberse a que no se realizan de forma rutinaria pruebas radiológicas por falta de protocolos bien establecidos. La atresia pilórica, producida por mutaciones en genes que codifican para la proteína integrina α6β4, aparece de forma precoz en formas de EBJ, al igual que lo encontrado en el 2,8% de nuestros pacientes17.
A nivel intestinal, el 43,9% de los pacientes presentó estreñimiento, siendo así el síntoma digestivo más frecuente encontrado en todas las formas de EB desde los primeros años de vida. Se ha estimado que ocurre en el 20 al 40% de los pacientes con EBS y EBJ, y en el 40 al 75% de los casos con EBDR16,18, coincidiendo con los hallazgos de nuestra serie.
La anemia multifactorial es frecuente en las formas graves de EB, apareciendo en el 36,4% de nuestros pacientes, por una combinación del déficit de hierro y de anemia por inflamación crónica19. El 35% de los casos presentaron retraso en el crecimiento, la mayoría con EBDR grave, coincidiendo con otras series reportadas anteriormente16.
La pseudosindactilia en manos y pies y las contracturas en flexión son complicaciones frecuentes, producidas por la aparición repetida de ampollas y cicatrices en los pacientes8. La pseudosindactilia se observa con mayor frecuencia en la EBDR, al igual que en nuestra cohorte. En una de las series más largas publicadas esta complicación aparecía a partir del primer año de edad (en el 13-16% de los pacientes con el subtipo EBDR grave), alcanzando niveles máximos del 98,2% en EBDR grave a los 20 años de edad20.
Las complicaciones oftalmológicas más frecuentes fueron las erosiones corneales y la queratitis, que aparecieron en el 22% de los casos totales, y en el 31,9% de pacientes con EBDR. Este dato es algo inferior al reportado en otras series7,21 al no estar bien documentado en la historia clínica en todos los casos, ya que algunos pacientes son valorados en otras comunidades.
Entre las complicaciones genitourinarias, cinco pacientes tuvieron glomerulonefritis y uno estenosis meatal, todos ellos con EBDR. En otras series reportadas, el riesgo acumulado de muerte por insuficiencia renal en pacientes con EBDR grave fue del 12,3% a los 35 años de edad22. En nuestra cohorte, dos pacientes con EBDR fallecieron en la segunda década de la vida, a consecuencia del fallo renal.
El daño en el aparato respiratorio se observa predominantemente en las formas con EBJ grave7. Ninguno de los pacientes incluidos en nuestra cohorte presentó manifestaciones de la esfera respiratoria, salvo una paciente con EBS-DM.
Las alteraciones cardíacas aparecieron en 5,6% de los pacientes. La miocardiopatía dilatada (MCD) es una complicación poco frecuente de la EBDR grave, pero puede llegar a ser mortal, especialmente en aquellos pacientes con insuficiencia renal crónica concurrente. Aunque en series anteriores se había observado un mayor riesgo acumulado de miocardiopatía en los pacientes con EBDR grave de mayor edad (4,5% a partir de los 20 años)23, en los últimos años han aparecido nuevos casos reportados a edades inferiores24, coincidiendo con los hallazgos de nuestra serie, en la que los tres pacientes con MCD grave eran menores de ocho años. Esto plantea la necesidad de realizar un despistaje precoz con ecocardiograma desde edades tempranas, sobre todo en formas graves de EB.
El CCE es una de las principales causas de mortalidad en estos pacientes, siendo más frecuente en las formas con EBDR, en las que el riesgo acumulado de desarrollo de CCE alcanza un 90,1% a los 55 años25. En nuestra serie, la edad mediana de aparición de CCE fue 26,50 años (RIQ: 20,00-31,00), similar a lo descrito en otras series8. A pesar de que una de las pacientes con EBS incluida en nuestra cohorte desarrolló un CCE, este tipo de tumor es infrecuente en este subtipo de EB26. Además, los CCE asociados con la EB son muy agresivos, independientemente de sus características histopatológicas, con altas tasas de recurrencia y mortalidad27.
En nuestra serie, un total de 22 pacientes han fallecido, siendo la sepsis y el CCE las causas más frecuentes. Las formas de EBJ grave son las que presentan un peor pronóstico, con una esperanza de vida habitualmente inferior a un año, tal y como aparece reflejado en la literatura28, y como se ha podido constatar en los tres pacientes incluidos en nuestra cohorte.
Entre las principales limitaciones del estudio debemos mencionar principalmente las propias de los estudios retrospectivos. Es posible que exista un infrarregistro de algunas complicaciones y que la falta de alguna variable pueda haber influido en los resultados finales. No se buscaron en todos los pacientes los problemas en la esfera psicológica mediante escalas específicas, habiéndose reportado altas tasas de síndrome ansioso-depresivo29. Tampoco se investigó la presencia de alteraciones óseas, a pesar de haberse descrito una alta frecuencia de osteopenia y osteoporosis en formas graves de EB30.
Como conclusiones, la forma clínica predominante en nuestra serie fue la EBDR. Las complicaciones más prevalentes fueron el prurito, el dolor y las infecciones cutáneas, y las más graves, la miocardiopatía y el CCE. La derivación y el seguimiento de estos pacientes en centros de referencia aseguran un adecuado manejo multidisciplinar, liderado por un dermatólogo con experiencia en EB.
A todos los profesionales que forman parte del equipo multidisciplinar que participa en la atención de los pacientes con EB y a la Asociación Piel de Mariposa DEBRA-España, por su apoyo incondicional.
A todos los investigadores que forman parte del grupo CIBERER-CIEMAT-UC3M-IISFJD (Departamento de Bioingeniería, Universidad Carlos III de Madrid; Centro de Investigación Biomédica en Red de Enfermedades Raras [U714-CIBERER]. Unidad de Medicina Regenerativa, CIEMAT; Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz) y del Servicio de Diagnóstico Genético del Instituto de Investigación de Enfermedades Raras; Instituto de Salud Carlos III.



