






La epidermólisis ampollosa adquirida es una enfermedad ampollosa subepidérmica autoinmune causada por autoanticuerpos contra el colágeno vii. Su clínica es heterogénea con afectación de la piel y las mucosas, pudiendo generar secuelas invalidantes. Existen diversas opciones terapéuticas frecuentemente insatisfactorias.
ObjetivoRevisar los casos de epidermólisis ampollosa adquirida diagnosticados durante un periodo de 26 años.
Material y métodosEstudio retrospectivo de las características clínicas e inmunopatológicas de 9 pacientes con dicho diagnóstico.
ResultadosLa mediana de edad de presentación fue de 37 años, el 66,67% de pacientes fueron mujeres. Asociaciones: neoplasias malignas, enfermedad inflamatoria intestinal y procesos autoinmunes. La variante inflamatoria fue la más frecuente (6/9). La histología mostró constantemente una ampolla subepidérmica y la inmunofluorescencia directa la presencia de depósitos lineales de IgG y C3 en la membrana basal. La inmunofluorescencia indirecta fue positiva en 6 pacientes, mostrando en todos ellos un patrón dérmico en piel separada. En 5 pacientes se determinaron los anticuerpos contra el colágeno vii por Enzyme-Linked Immuno Sorbent Assay, de los cuales 2 fueron positivos, e Inmunoblot con NC1 recombinante en 6 casos, positivo en todos ellos. La respuesta terapéutica fue variable.
ConclusionesSe trata de una enfermedad rara, de clínica heterogénea, que puede inducir a confusión con otras enfermedades ampollosas subepidérmicas. Se requiere un alto índice de sospecha y el empleo de todos los métodos disponibles para establecer su diagnóstico. La correcta evaluación de la afectación cutáneo-mucosa y la instauración precoz de la terapéutica adecuada permitirá la detección de sus secuelas y de las complicaciones del tratamiento.
Epidermolysis bullosa acquisita (EBA) is an autoimmune subepidermal blistering disease caused by autoantibodies to type VII collagen. The clinical presentation is variable, with skin and mucosal lesions that can cause significant dysfunction. Different treatment options exist, but the results are often unsatisfactory.
ObjectiveTo review all the cases of epidermolysis bullosa acquisita (EBA) diagnosed at our hospital over a 26-year period.
Materials and methodsWe performed a retrospective review of the clinical, histologic, and immunologic features of EBA in 9 patients.
ResultsMean age at presentation was 37 years and 66.67% of the patients were women. EBA occurred in association with malignant tumors, inflammatory bowel disease, and autoimmune disorders. The most common variant was inflammatory EBA (6 of the 9 cases). In all 9 patients, histology revealed a subepidermal blister and direct immunofluorescence showed linear deposits of immunoglobulin G and C3 in the basement membrane zone. Indirect immunofluorescence performed on salt-split skin substrate was positive in 6 patients and showed a dermal pattern in all cases. Five patients were tested for autoantibodies to type VII collagen using enzyme-linked immunosorbent assay, with positive results in 2 cases. Immunoblotting using recombinant noncollagenous domains (NC1) of type VII collagen was positive in all 6 cases in which it was performed. Response to treatment was variable.
ConclusionsEBA is a rare disease with a variable clinical presentation that can be confused with that of other subepidermal blistering diseases. Correct diagnosis requires a high level of clinical suspicion and the use of all available diagnostic tests. Thorough evaluation of cutaneous and mucosal involvement and prompt initiation of appropriate treatment will ensure the detection and prevention of dysfunction and treatment-related complications.
La epidermólisis ampollosa adquirida (EAA) es una enfermedad ampollosa subepidérmica crónica que afecta la piel y mucosas1–3. Fue descrita originalmente por Roenigk et al.1, en 1971 como un cuadro mecano-ampolloso (asociado a fragilidad cutánea), semejante a la epidermólisis ampollosa congénita, pero de inicio en la edad adulta y sin antecedentes familiares. En 1973 Kushniruk et al.4 describen la presencia de depósitos de IgG y C3 en la membrana basal en el examen de inmunofluorescencia directa (IFD) en la piel de estos pacientes. Yaoita et al.5 en 1981 demostraron por inmunomicroscopia electrónica la localización de los depósitos en la sublámina densa en la EAA y en la lámina lúcida en el penfigoide ampolloso. En 1984 Woodley et al.6 describen una proteína de 290 KDa como la diana antigénica en la EAA, que posteriormente se identificó como colágeno vii7. Posteriormente se describió una variedad inflamatoria con características clínicas similares al penfigoide ampolloso y otros subtipos que remedaban el penfigoide de mucosas o el penfigoide cicatricial de Brunsting-Perry8–10. Durante el curso de la enfermedad puede haber una transformación de una presentación clínica a otra o coexistir rasgos de ambas11.
Se trata de una enfermedad poco frecuente, con una incidencia estimada de 0,2 casos/millón de habitantes2,3. Debido a la baja prevalencia no se ha podido establecer si existe predilección racial, aunque se ha documentado una mayor prevalencia en individuos de raza negra y en coreanos12,13. La edad de inicio suele ser entre los 40 y 50 años, aunque se han descrito casos en ancianos, en niños, así como un caso congénito por trasmisión vertical de una madre afecta14–16.
La etiología de la EAA es desconocida, pero el papel de los anticuerpos contra el colágeno vii en el desarrollo de la enfermedad parece evidente. La patogenicidad de estos anticuerpos ha sido demostrada por la aparición de ampollas en una recién nacida por la transferencia materno-fetal de dichos anticuerpos a partir de la sangre de su madre afecta de EAA. También se ha podido evidenciar ex-vivo: la incubación de criosecciones de piel humana con el suero de pacientes con EAA, en presencia de neutrófilos, produce una separación dermoepidérmica. El desarrollo de diferentes modelos in vivo mediante inmunización pasiva (transferencia de IgG anti-colágeno vii humano o de conejo a ratones) o activa (inmunización de determinadas estirpes de ratones con fragmentos inmunodominantes del NC1 del colágeno vii) ha permitido producir formas de EAA experimental16–19.
Histológicamente, las formas de EAA clásicas mecano-ampollosas se caracterizan por la presencia de ampollas subepidérmicas con escaso infiltrado inflamatorio. En las formas de EAA inflamatoria los infiltrados inflamatorios son de mayor intensidad, con la presencia de abundantes neutrófilos y en ocasiones de eosinófilos. La IFD muestra depósitos lineales, predominantemente de IgG, a lo largo de la membrana basal dermoepidérmica. En algunos pacientes también se pueden observar depósitos de C3, IgA, o IgM de intensidad variable4,5,20. El examen con inmunofluorescencia indirecta (IFI) permite detectar, en algunos pacientes, la presencia de autoanticuerpos circulantes en el suero de tipo IgG dirigidos contra la membrana basal dermoepidérmica. Se pueden emplear diferentes sustratos, como la piel humana o el esófago de mono5, pero los mejores resultados se obtienen empleando piel humana separada con cloruro sódico (NaCl) 1M. Con esta última técnica se puede demostrar la unión de los autoanticuerpos en la EAA a la porción dérmica (suelo) de la separación, a diferencia de lo que sucede en el penfigoide ampolloso, donde se unen a la porción epidérmica de la separación. Aunque este método relativamente sencillo permite diferenciar la EAA del penfigoide ampolloso, no la diferencia de otras enfermedades ampollosas autoinmunes subepidérmicas con patrón dérmico (lupus eritematoso ampolloso, penfigoide anti-laminina-332, penfigoide anti-γ1-laminina o penfigoide p: anti p105)21. En aquellos casos en los que la IFI es negativa se puede realizar una IFD previa separación de la biopsia con NaCl 1M (o en biopsia de una ampolla provocada por succión). Dicha técnica muestra la localización de los depósitos de IgG en la porción dérmica de la membrana basal separada en la EAA, pero al igual que la IFI no permite la diferenciación de otras enfermedades con patrón dérmico22. Recientemente se ha descrito también el valor diagnóstico del patrón morfológico de los depósitos de anticuerpos en la membrana basal. En el caso de que los depósitos de IgG estén por encima de la sublámina densa (penfigoide ampolloso, penfigoide anti-laminina-332) se observa un patrón serrado con configuración en n (n-serrado). Si los depósitos se sitúan en la sublámina densa (EAA) el patrón serrado adopta una configuración en u (u-serrado). Esta técnica requiere que los cortes sean de menos de 4μm de grosor23.
Cuando se realiza el análisis del suero de los pacientes con EAA mediante técnicas de inmunoblot utilizando extractos dérmicos, estos identifican unas bandas proteicas de aproximadamente 290-kDA y 145-kDA, correspondientes al colágeno vii completo (dímero) o a una de sus regiones (monómero). La mayoría de los epítopos antigénicos del colágeno vii se localizan en la porción NC1 (de 145-kDA). Por dicho motivo se puede emplear también la proteína recombinante NC1 producida en el laboratorio para realizar el análisis por Inmunoblot, o mediante la técnica de ELISA. La ELISA con NC1 recombinante es bastante más sensible y específica que la IFI con piel separada o el Inmunoblot con extractos dérmicos, y permite además cuantificar la cantidad de anticuerpos24. Recientemente se ha desarrollado una técnica con colágeno vii total (que incluye tanto la región NC1, como la NC2) que es ligeramente más sensible que la técnica con NC125.
El método diagnóstico considerado idóneo es la microscopia inmunoelectrónica, dado que permite identificar la localización de los depósitos de inmunoglobulinas: en la sublámina densa en la EAA y en la lámina lúcida y en los hemidesmosomas en el penfigoide ampolloso5,20. Sin embargo, se trata de una técnica complicada, disponible en muy pocos centros.
La correlación de la clínica con el resultado de los diferentes métodos diagnósticos permite establecer con cierta seguridad el diagnóstico de EAA, pero no diferenciarlo del lupus eritematoso sistémico ampolloso (LESA), ya que ambas enfermedades presentan anticuerpos patógenos frente al colágeno vii26. En el LESA deben cumplirse los criterios de LES del American College of Rheumatology, las lesiones predominan en zonas fotoexpuestas y su curso es menos refractario que el de la EAA27.
Por lo que respecta al tratamiento de la EAA se han empleado diversos tratamientos frecuentemente ineficaces y de respuesta imprevisible, existen pocas series largas de pacientes y no hay estudios aleatorizados en esta enfermedad dada su infrecuencia3,28.
En el presente trabajo hemos analizado las características demográficas, clínicas, inmunopatológicas, así como la respuesta terapéutica de una serie de pacientes con EAA diagnosticados durante el periodo comprendido entre los años 1985 y 2011 en nuestra institución.
Pacientes y métodosSe estudiaron retrospectivamente todos los pacientes diagnosticados de EAA en el Hospital Clínic de Barcelona en el periodo comprendido entre los años 1985 y 2011 en función de criterios clínicos, histológicos e inmunopatológicos. Se analizaron todos los datos demográficos, clínicos, inmunopatológicos y terapéuticos disponibles.
La IFI empleando piel humana separada NaCl 1M, según la técnica convencional, se realizó utilizando el suero obtenido en una fase de actividad21. La IFD con piel separada se practicó según la técnica descrita por Gammon22. Los estudios de Inmunoblot se realizaron utilizando extractos dérmicos y en algunos casos también proteínas recombinantes codificando para la fracción NC1 del colágeno vii. La técnica de ELISA se realizó utilizando diferentes moléculas recombinantes del colágeno vii (His-hCVII-NC1 y NC2-His-hCVII-NC1 NC2-H) según el procedimiento descrito por los autores25.
ResultadosIdentificamos 9 pacientes con diagnóstico de EAA durante el periodo 1985-2011. La mediana de edad de presentación fue de 37 años, 3 varones y 6 mujeres (relación hombre:mujer de 1:2). La mediana de tiempo transcurrido hasta el diagnóstico fue de 7 meses (rango de 1 a 72 meses).
La forma de presentación más frecuentemente observada fue la inflamatoria (6/9) (fig. 1). La variedad clásica mecano-ampollosa ocurrió en los pacientes de edad más avanzada (todos ellos mayores de 72 años) (fig. 2). En un paciente se produjo un cambio de fenotipo clínico de penfigoide ampolloso a penfigoide de Brunsting-Perry (caso 5 [tabla 1]). Solo en 2 casos la afectación fue exclusivamente cutánea. De los 7 pacientes con compromiso de mucosas todos mostraron afectación oral, 2 de ellos genital, 2 conjuntival y un paciente afectación faríngea, laríngea y esofágica.
, ampolla subepidérmica. C. Hematoxilina-eosina (×100), escaso infiltrado inflamatorio.")
: ampolla subepidérmica. C. Hematoxilina-eosina (×100): infiltrado inflamatorio importante con predominio de neutrófilos y escasos eosinófilos.")
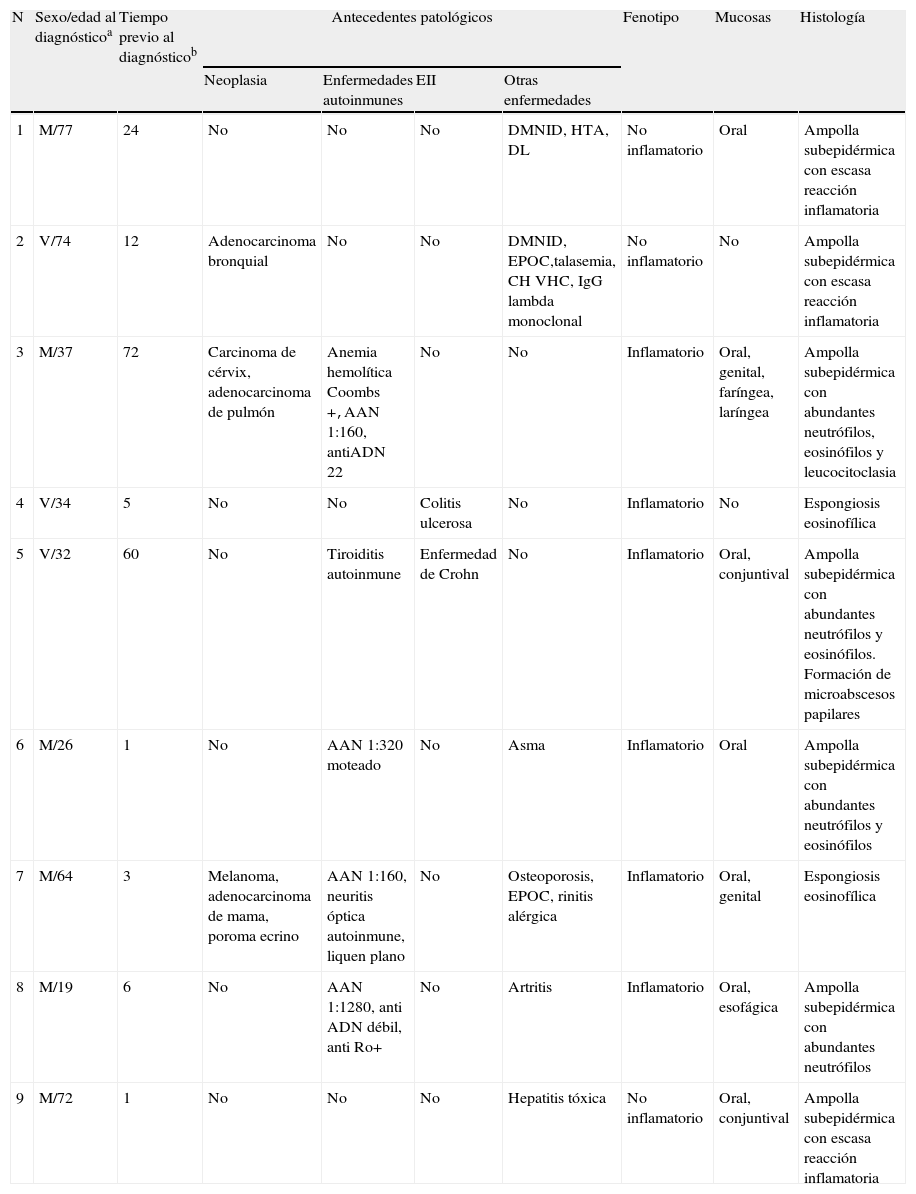
Características clínicas e inmunopatológicas de los pacientes con epidermólisis ampollosa adquirida
| N | Sexo/edad al diagnósticoa | Tiempo previo al diagnósticob | Antecedentes patológicos | Fenotipo | Mucosas | Histología | |||
| Neoplasia | Enfermedades autoinmunes | EII | Otras enfermedades | ||||||
| 1 | M/77 | 24 | No | No | No | DMNID, HTA, DL | No inflamatorio | Oral | Ampolla subepidérmica con escasa reacción inflamatoria |
| 2 | V/74 | 12 | Adenocarcinoma bronquial | No | No | DMNID, EPOC,talasemia, CH VHC, IgG lambda monoclonal | No inflamatorio | No | Ampolla subepidérmica con escasa reacción inflamatoria |
| 3 | M/37 | 72 | Carcinoma de cérvix, adenocarcinoma de pulmón | Anemia hemolítica Coombs+,AAN 1:160, antiADN 22 | No | No | Inflamatorio | Oral, genital, faríngea, laríngea | Ampolla subepidérmica con abundantes neutrófilos, eosinófilos y leucocitoclasia |
| 4 | V/34 | 5 | No | No | Colitis ulcerosa | No | Inflamatorio | No | Espongiosis eosinofílica |
| 5 | V/32 | 60 | No | Tiroiditis autoinmune | Enfermedad de Crohn | No | Inflamatorio | Oral, conjuntival | Ampolla subepidérmica con abundantes neutrófilos y eosinófilos. Formación de microabscesos papilares |
| 6 | M/26 | 1 | No | AAN 1:320 moteado | No | Asma | Inflamatorio | Oral | Ampolla subepidérmica con abundantes neutrófilos y eosinófilos |
| 7 | M/64 | 3 | Melanoma, adenocarcinoma de mama, poroma ecrino | AAN 1:160, neuritis óptica autoinmune, liquen plano | No | Osteoporosis, EPOC, rinitis alérgica | Inflamatorio | Oral, genital | Espongiosis eosinofílica |
| 8 | M/19 | 6 | No | AAN 1:1280, anti ADN débil, anti Ro+ | No | Artritis | Inflamatorio | Oral, esofágica | Ampolla subepidérmica con abundantes neutrófilos |
| 9 | M/72 | 1 | No | No | No | Hepatitis tóxica | No inflamatorio | Oral, conjuntival | Ampolla subepidérmica con escasa reacción inflamatoria |
| N | Inmunofluorescencias | ELISA col vii | Inmunoblot | ||
| IFD | IFD piel separada | IFI piel separada | |||
| 1 | IgG, C3 | NR | Dérmico,1:320 | NR | Laminina 332 (IgG4)- negativo, Colágeno vii (NC1 para IgG1, IgG3, IgG4)-positivo |
| 2 | IgG, IgM, C3 | Dérmico | Negativa | Negativo | Anti laminina 332 (IgG4)- negativo, Colágeno vii (NC1 para IgG1, IgG3, IgG4)-positivo |
| 3 | IgG, IgA, C3 | NR | Dérmico,IgA e IgG 1:320 | Positivo | NC1: IgG4 ++ IgA positivo |
| 4 | IgG, C3 | Dérmico | Negativa | NR | Laminina 332 (IgG4)- negativo Colágeno vii (NC1 para IgG1, IgG3, IgG4)-positivo |
| 5 | IgG, IgA, C3 | Dérmico | Dérmico, 1:160 | Negativo | Laminina 332- negativo, Colágeno vii (NC1 para IgG1, IgG3, IgG4)-negativo |
| 6 | IgG, IgA, C3 | Dérmico | NR | NR | NR |
| 7 | IgG, C3 | Dérmico | Dérmico, 1:20 | Positivo | NR |
| 8 | IgG, IgM, IgA, C3, fibrinógeno | Dérmico | Dérmico, 1:20 (AAN dermoepidérmico) | Negativo | NC1:IgG1-IgG4-IgA Colágeno vii positivo |
| 9 | IgG, C3 | NR | Dérmico | NR | NR |
AAN: anticuerpos antinucleares; CH por VHC: cirrosis hepática por virus de hepatitis C; DL: dislipidemia; DMNID: diabetes mellitus no insulinodependiente; EII: enfermedad inflamatoria intestinal; ELISA col VII: enzime-linked immunoabsorbent assay for collagen VII; EPOC: enfermedad pulmonar obstructiva crónica; HTA: hipertensión arterial. IFD: inmunofluorescencia directa; IFI: inmunofluorescencia indirecta; NR: no realizado.
aAño; bMeses.
Al revisar la asociación con otras enfermedades y procesos destacó una asociación frecuente a neoplasias malignas (5 neoplasias en 3 pacientes: 2 de pulmón, un carcinoma de cérvix, uno de mama y un melanoma maligno), enfermedad inflamatoria intestinal (EII) (una enfermedad de Crohn y una colitis ulcerosa), así como procesos autoinmunes o autoanticuerpos circulantes (una anemia hemolítica, una neuritis óptica, una tiroiditis, 4 anticuerpos antinucleares [AAN] positivos) (tabla 1).
En todos los casos de EAA con la forma clásica el examen histológico mostró una ampolla subepidérmica con infiltrado inflamatorio muy escaso. En los 6 pacientes con clínica inflamatoria se apreció un infiltrado formado por abundantes neutrófilos y eosinófilos, con presencia ocasional de microabscesos en la dermis papilar. En 2 casos se observó espongiosis eosinofílica. En el estudio de IFD fue constante la presencia de depósitos lineales de IgG y de C3 en la membrana basal, de intensidad variable (fig. 3). En 5 (56%) existían además depósitos de otros conjugados (tabla 1). En 8 de los pacientes se realizó la IFI en piel humana separada con NaCl 1M, resultando positiva con patrón dérmico en 6 de ellos (75%) (fig. 4). En uno de los pacientes (caso 6 [tabla 1]) el examen de IFI se realizó en otro centro, mostrando exclusivamente la presencia de ANA, y no dispusimos de suero para realizar el estudio en piel separada. La IFD con piel separada se practicó en 6 de los pacientes mostrando en todos ellos (incluyendo los 2 casos con IFI negativa y la paciente en la que no se practicó estudio de IFI), un patrón exclusivamente dérmico de los depósitos de IgG.
, depósitos lineales intensos de IgG (A) y muy tenues de C3 (B) a lo largo de la unión dermoepidérmica.")
, los anticuerpos están dirigidos contra la parte dérmica (suelo de la ampolla).")
En 5 pacientes se realizó ELISA para colágeno vii, siendo positivo en 2 (40%). En 6 pacientes se practicó estudio de Inmunoblot resultando positivo en 5 (83%) de ellos (tabla 1) (fig. 5). Se detectó la presencia de ANA en 4 pacientes (44%), en una de ellas como hallazgo aislado (caso 6 [tabla 1]), en 2 en asociación con otras enfermedades autoinmunes (casos 3 y 7 [tabla 1]). La paciente 8 (tabla 1), a pesar de tener ANA y anti-ADN positivos, no cumplió en ningún momento de su evolución los criterios de LES de la American College of Rheumatology. En esta paciente, además, la falta de respuesta a corticoides y dapsona a dosis altas y la buena respuesta al tratamiento con inmunoglobulina intravenosa (IGIV) asociada a colchicina, nos inclinó a considerar el diagnóstico de EAA más que el de LESA, si bien ambos procesos tienen características inmunológicas semejantes y existe un cierto solapamiento.
. Se observa una banda de 145kD (correspondiente a la región NC1 del colágeno vii). CN: control negativo; CP: control positivo; P: suero paciente.")
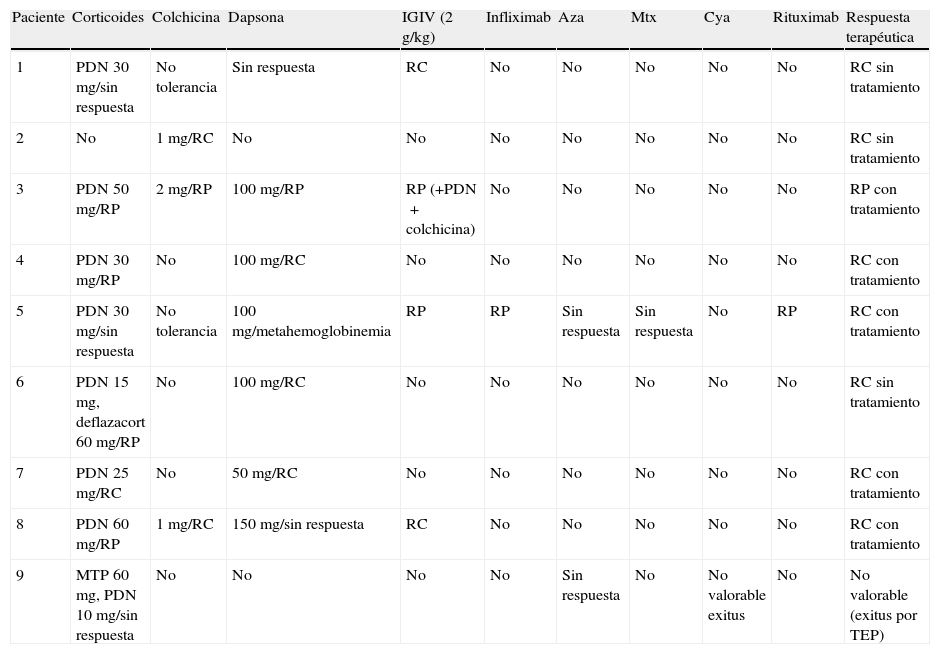
Se realizaron diversos tratamientos desde el momento del diagnóstico con respuesta clínica variable en la mayoría de los pacientes y difícil control de la enfermedad en 2 casos (casos 3 y 5 [tabla 2]). La paciente número 3 inició tratamiento con prednisona a dosis de 1mg/kg/día con respuesta parcial, se añadió colchicina 1mg/12h y posteriormente dapsona 100mg/día con poco resultado, por lo que se inició tratamiento con IGIV a dosis altas (2g/kg), y ante la falta de respuesta se añadió prednisona y colchicina con control parcial de la enfermedad. En el curso de su enfermedad se detectó un adenocarcinoma pulmonar con posterior desarrollo de metástasis y desenlace fatal. El caso número 5, el más refractario de todos, realizó diversos tratamientos suspendidos bien por efectos adversos bien por falta de respuesta; en diciembre de 2007 recibió tratamiento con rituximab (375mg/m2/semana, 4 semanas) con buena respuesta cutánea, pero persistiendo la gingivitis erosiva. Actualmente se encuentra en remisión completa con dosis mínimas de corticoides, dapsona y colchicina. Destacamos la excelente respuesta a las IGIV de una paciente con la forma clásica que fue refractaria a todos los tratamientos previos (caso 1 [tabla 2]).
Tratamientos/respuesta
| Paciente | Corticoides | Colchicina | Dapsona | IGIV (2g/kg) | Infliximab | Aza | Mtx | Cya | Rituximab | Respuesta terapéutica |
| 1 | PDN 30mg/sin respuesta | No tolerancia | Sin respuesta | RC | No | No | No | No | No | RC sin tratamiento |
| 2 | No | 1mg/RC | No | No | No | No | No | No | No | RC sin tratamiento |
| 3 | PDN 50mg/RP | 2mg/RP | 100mg/RP | RP (+PDN+colchicina) | No | No | No | No | No | RP con tratamiento |
| 4 | PDN 30mg/RP | No | 100mg/RC | No | No | No | No | No | No | RC con tratamiento |
| 5 | PDN 30mg/sin respuesta | No tolerancia | 100mg/metahemoglobinemia | RP | RP | Sin respuesta | Sin respuesta | No | RP | RC con tratamiento |
| 6 | PDN 15mg, deflazacort 60mg/RP | No | 100mg/RC | No | No | No | No | No | No | RC sin tratamiento |
| 7 | PDN 25mg/RC | No | 50mg/RC | No | No | No | No | No | No | RC con tratamiento |
| 8 | PDN 60mg/RP | 1mg/RC | 150mg/sin respuesta | RC | No | No | No | No | No | RC con tratamiento |
| 9 | MTP 60mg, PDN 10mg/sin respuesta | No | No | No | No | Sin respuesta | No | No valorable exitus | No | No valorable (exitus por TEP) |
AZA: azatioprina; CyA: ciclosporina; IGIV: inmunoglobulinas intravenosas; MTP: metilprednisolona; MTX: metotrexato; PDN: prednisona; RC (respuesta completa) sin tratamiento: ausencia de lesiones en un periodo mínimo de 2 meses; RC con tratamiento: ausencia de lesiones con tratamiento mínimo (igual o equivalente a 8mg/d metilprednisolona con o sin dapsona o colchicina por mínimo de 2 meses); RP (respuesta parcial) sin tratamiento: lesiones nuevas transitorias que curan en una semana sin tratamiento por un mínimo de 2 meses; RP con tratamiento mínimo: lesiones nuevas transitorias que curan en una semana en paciente con tratamiento mínimo. TEP: tromboembolismo pulmonar.
La EAA es una enfermedad poco frecuente que afecta predominantemente a adultos1–3. En nuestro estudio la mediana de edad en el momento del diagnóstico fue de 37 años, y encontramos un predominio en mujeres, al igual que en las 4 únicas series publicadas29–32.
La forma clásica mecano-ampollosa corresponde a la que se reportó en la descripción original de la EAA. Tiene un fenotipo bastante característico que facilita la orientación diagnóstica; sin duda por ello la mayoría de los casos inicialmente publicados correspondían a esta presentación clínica. Desde la descripción de la variedad inflamatoria se ha documentado un incremento progresivo de casos diagnosticados de esta forma. Posiblemente el mayor conocimiento de la EAA y el empleo adecuado de los métodos diagnósticos disponibles ante la sospecha clínica ha permitido establecer este diagnóstico en pacientes que de otra forma serían diagnosticados de penfigoide ampolloso. La variedad clínica más frecuente en nuestra serie fue la inflamatoria, al igual que en las series recientemente publicadas. Sin embargo, hemos observado un predominio de la forma clásica en los pacientes de edad mas avanzada, lo que no se ha descrito en los otros estudios29–32. No tenemos una explicación para este hallazgo y el bajo número de pacientes no permite obtener conclusiones al respecto. La asociación de la EAA con otras enfermedades es bien conocida, siendo probablemente la más establecida la EII, y en particular la enfermedad de Crohn1,3. La presencia de colágeno vii en el epitelio intestinal podría explicar esta asociación como un fenómeno de expansión de epítopos, hipótesis apoyada por la presencia de anticuerpos circulantes contra el colágeno vii en aproximadamente un 68% de pacientes con enfermedad de Crohn (aunque no presenten enfermedad cutánea)33,34. En nuestra serie la incidencia de neoplasias fue superior a la de EII. Se han descrito casos aislados asociados a diferentes neoplasias, sin embargo ninguna de las series publicadas hasta ahora hace referencia a esta asociación. El curso de la enfermedad en nuestros pacientes no parece sugerir un proceso paraneoplásico. De hecho, las 3 neoplasias previamente conocidas estaban en remisión en el momento del inicio de la EAA. En la paciente 3 hubo un retraso diagnóstico de 72 meses, detectándose la neoplasia en el momento del establecimiento del diagnóstico en nuestro hospital; no recibió fármacos inmunosupresores que pudieran actuar como desencadenante. En el paciente 2 la neoplasia comenzó tras la remisión completa de la EAA y había sido tratado exclusivamente con colchicina.
Con respecto al examen histológico, tal como se ha documentado en la literatura, el grado de inflamación clínica se correlacionó con la intensidad del infiltrado inflamatorio. La IFD no permitió el diagnóstico diferencial de la EAA con el penfigoide ampolloso, a pesar de que, tal como está descrito, observamos a menudo una mayor intensidad de los depósitos de IgG respecto a los de C3, a diferencia de lo que sucede en el penfigoide, en el que los depósitos de C3 son más intensos35. La IFD con piel separada fue el método diagnóstico de mayor sensibilidad, mostrando un patrón dérmico en todos los pacientes estudiados. La IFI con piel separada fue negativa solo en 2 pacientes. En cuanto a los patrones morfológicos de inmunodepósito en IFD, serrados en «n» o en «u», no los pudimos valorar en nuestra serie, posiblemente debido a problemas técnicos como el grosor de las secciones. Es necesario comentar que en el caso 5 las pruebas realizadas no permiten una confirmación diagnóstica definitiva de EAA. La negatividad de los anticuerpos anti-BP 180, la afectación mucosa, la asociación con enfermedad de Crohn y el curso tórpido y refractario a los tratamientos sugieren un diagnóstico probable de EAA, aunque no descartamos que se trate de una EAA por IgA por no haber podido realizar ELISA para anticuerpos IgA, si bien el paciente mostraba depósitos de IgG en la membrana basal mucho más intensos que los de IgA y de C3.
La respuesta terapéutica fue muy variable y no siempre satisfactoria. Destacamos el excelente resultado de las IGIV en una paciente con una forma clásica que había sido refractaria a múltiples tratamientos previos (tabla 2). Basándonos en nuestra experiencia, nuestra propuesta terapéutica para la EAA sería iniciar prednisona 0,5mg/kg (o equivalente) asociando colchicina 1-2mg/día y/o dapsona 25-100mg/día. Si la respuesta permite la disminución de la dosis de prednisona hasta 5mg (o su supresión) se mantendría esta medicación. En caso de no poder disminuir la corticoterapia (a pesar de administrar dosis de hasta 100mg de dapsona y 2mg de colchicina) sugerimos añadir a dicho tratamiento las IGIV. Actualmente el uso de rituximab parece mostrar una eficacia superior (y un coste menor), por lo que en casos refractarios podría plantearse como una buena opción, si bien nuestra experiencia con este fármaco en EAA se limita a un solo paciente.
En resumen, presentamos la primera serie de EAA en España. Al igual que en las 4 únicas series publicadas incluyendo 38, 30, 14 y 12 casos29–32, observamos un predominio de las formas inflamatorias. Es importante en primer lugar la sospecha clínica y la optimización de los métodos diagnósticos disponibles que permitan establecer un diagnóstico de certeza. Lehman et al.36 proponen un algoritmo diagnóstico en enfermedades ampollosas autoinmunes, con valoración del coste de las diversas pruebas y el empleo racional de las mismas. Una vez establecido el diagnóstico debe realizarse una evaluación exhaustiva de la extensión de la enfermedad y detección de posibles procesos patológicos asociados, e iniciar un tratamiento adecuado que permita la prevención de sus secuelas en aquellos casos con afectación ocular, de mucosa respiratoria o digestiva. La respuesta terapéutica a un fármaco determinado es poco valorable por tratarse de una enfermedad muy heterogénea.
Se trata de un estudio retrospectivo que incluye un número bajo de pacientes. Es necesario realizar estudios prospectivos multicéntricos que permitan un mejor conocimiento de los mecanismos implicados en esta enfermedad, de la eficacia de los diversos fármacos y establecer la individualización del tratamiento.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.
Al Dr. C. Sitaru por la realización de ELISA para colágeno vii. Al Dr. J. Herrero y al Dr. A. Guilabert por la realización de los Inmunoblots.



