







INTRODUCCION
Las mucopolisacaridosis son un conjunto de enfermedades de depósito en las que el déficit de una enzima lisosomal ocasiona la acumulación de determinados mucopolisacáridos produciendo diversas manifestaciones clínicas1. En general se trata de alteraciones esqueléticas, corneales, neurológicas y viscerales, aunque también pueden encontrarse manifestaciones cutáneas. La enfermedad de Hunter o mucopolisacaridosis tipo II está ocasionada por el déficit de la enzima iduronato 2 sulfatasa, siendo el dermatán y heparán sulfato los mucopolisacáridos depositados. La aparición de lesiones papulares simétricas «en empedrado» en la región escapular y caras laterales externas de brazos y muslos se ha considerado patognomónica de este síndrome2,3. Presentamos el caso de un varón de 7 años que fue diagnosticado de mucopolisacaridosis II o síndrome de Hunter en su forma moderada con lesiones cutáneas características.
DESCRIPCION DEL CASO
Un varón de 7 años, con antecedentes de esplenomegalia e insuficiencia mitral, fue diagnosticado de mucopolisacaridosis tipo II o síndrome de Hunter. El paciente tenía concentraciones disminuidas de iduronato 2 sulfatasa en plasma y aumento de dermatán y heparán sulfato en orina. Al estudiar a la familia se comprobó que su hermano mayor y dos primos hermanos también estaban afectados, con niveles de iduronato 2 sulfatasa en plasma por debajo de los normales y aumento de dermatán y heparán sulfato en orina. No se realizó estudio genético.
El paciente acudió a consulta por la aparición en los últimos 6 meses de unas lesiones papulares asintomáticas en cara externa de brazos y muslos. En la exploración el niño presentaba una facies tosca, con el puente nasal deprimido, frente prominente y orejas de implantación baja (fig. 1). Otros hallazgos fueron cuello corto, macrocefalia y tórax en quilla. No existía retraso psicomotor ni del crecimiento. Las manos tenían un característico aspecto «en garra» (fig. 2), secundario a la contractura en flexión de las articulaciones interfalángicas distales. En cara externa de brazos y muslos se observaron unas lesiones papulares de 0,5 a 1 cm de diámetro, hipopigmentadas y agrupadas con aspecto «en empedrado» formando una placa (figs. 3 y 4). La histología de una de estas lesiones mostraba depósitos de una sustancia basófila entre los haces de colágeno de la dermis reticular con ausencia de infiltrados inflamatorios y de proliferación fibroblástica (fig. 5). Esta sustancia se teñía con hierro coloidal (fig. 6). Los hallazgos fueron compatibles con depósitos de mucopolisacáridos.
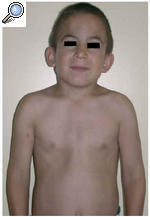
Fig. 1.--Aspecto clínico del paciente que presenta macrocefalia, cuello corto, orejas de implantación baja, facies tosca y tórax en quilla.
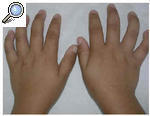
Fig. 2.--Manos «en garra» por contractura en flexión de las articulaciones interfalángicas distales.
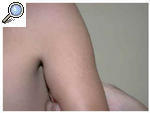
Fig. 3.--Pápulas hipopigmentadas de 0,5 a 1 cm de diámetro que confluyen dando aspecto «en empedrado» localizadas en la cara externa de ambos brazos.
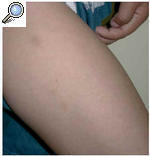
Fig. 4.--Imagen «en empedrado» en cara externa de ambos muslos por confluencia de pápulas hipopigmentadas idénticas a las que aparecen en los brazos.
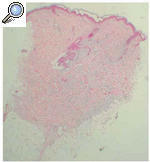
Fig. 5.--Depósito de una sustancia basófila entre los haces de colágeno de la dermis reticular en ausencia de infiltrados inflamatorios (Hematoxilina-eosina, *??.)
Fig. 6.--Positividad de la sustancia depositada entre los haces de colágeno con la tinción de hierro coloidal.
DISCUSION
Hunter describió este síndrome por primera vez en dos hermanos que presentaban estatura baja, contracturas articulares, manos «en garra», sordera, hepatosplenomegalia, cardiomegalia, facies tosca y unas lesiones nodulares sobre la región escapular4. Con los años se consiguió identificar este síndrome como una enfermedad de depósito lisosomal, en concreto la mucopolisacaridosis tipo II.
La mucopolisacaridosis tipo II se hereda de forma recesiva ligada a X a diferencia del resto de mucopolisacaridosis que son de herencia autosómica recesiva. La acumulación de heparán y dermatán sulfato secundario al déficit de iduronato 2 sulfatasa caracteriza a este síndrome.
Su incidencia es de 1/130.000 recién nacidos varones, aunque existe algún caso descrito en mujeres5.
El defecto se encuentra en un gen localizado en 27-28qX. No existe una exacta correlación entre genotipo y fenotipo, pero sí se sabe que, en general, las deleciones y los reordenamientos grandes dan lugar a fenotipos más graves. En España la mutación más frecuente es la G374sp, que se corresponde con un fenotipo moderado6.
Las manifestaciones cutáneas del síndrome de Hunter son variadas. Existen manifestaciones inespecíficas comunes a todas las mucopolisacaridosis1 como son cabello áspero, hipertricosis y un aspecto esclerodermiforme en el dorso de las manos con piel indurada y brillante. En cuanto a las manifestaciones específicas2,3 se trata de lesiones papulosas de 2 a 10 mm de diámetro de color piel normal o marfileño. A veces confluyen dando un aspecto reticulado o lineal. Se localizan preferentemente en la región escapular y las caras laterales de brazos y muslos, aunque también pueden aparecer en el tórax o la nuca. Las lesiones son asintomáticas, se inician antes de los 10 años de edad y tienden a desaparecer. Clásicamente se consideraban patognomónicas del síndrome de Hunter, aunque también se han descrito en otras mucopolisacaridosis. Aparecen tanto en la forma grave como en la moderada, de modo que su aparición no va a servir de indicador pronóstico.
La histopatología de estas lesiones muestra, en la dermis reticular, el depósito de una sustancia basófila entre los haces de colágeno que tiñe con hierro coloidal4. Esta sustancia corresponde a depósitos extracelulares de ácido hialurónico, dermatán y heparán sulfato. En la microscopia electrónica aparecen cuerpos residuales laminados electrodensos en fibroblastos, células de Schwann y macrófagos.
En cuanto al tratamiento, la compañía biotecnológica Transkaryotic therapies-TKT



