

INTRODUCCION
Las displasias ectodérmicas son un amplio y complejo grupo de trastornos hereditarios que fueron descritos por primera vez por Thurman en 18481. Estas entidades se caracterizan por un desarrollo anormal del tejido embrionario destinado a la formación de pelo, dientes, uñas, glándulas sudoríparas y otras estructuras ectodérmicas como las glándulas mamaria y lagrimal, el timo, el tiroides o el paladar. Por una serie de eventos complejos y poco conocidos, se producen ciertas anomalías en la unión epiteliomesenquimatosa que ocasionan una interrupción del desarrollo y dan lugar a la formación de estructuras ectodérmicas anormales.
Existen más de 170 cuadros diferentes, muchos de ellos descritos como casos aislados o en varios miembros de una misma familia, y algunos de ellos presentan además anomalías no relacionadas con estructuras ectodérmicas2. Clásicamente se ha utilizado una clasificación fenotípica2, aunque más recientemente Priolo y Laganà3 propusieron una nueva clasificación clinicogenética. Estos autores consideran que aunque sólo se conoce la alteración genética responsable de un pequeño número de estas enfermedades, se podría sospechar cuál es en relación con la sintomatología que presente el paciente.
Nosotros presentamos el caso de una niña de 4 años que presenta alteraciones compatibles con el síndrome AEC (anquilobléfaron, displasia ectodérmica y hendidura-cleft) o de Hay-Wells.
DESCRIPCION DEL CASO
Una niña de 4 años de edad, sin antecedentes familiares de interés, nacida a término tras un embarazo y parto sin complicaciones presentaba, en el momento del nacimiento, anquilobléfaron bilateral y hendidura palatina, trastornos que habían sido corregidos quirúrgicamente antes de acudir a nuestro servicio. Posteriormente desarrolló otros trastornos como piel seca y descamativa, fotofobia, intolerancia al calor sin cuadros de hiperpirexia, retraso en la dentición con anomalías dentarias y erosión e infecciones de cuero cabelludo y grandes pliegues que precisaban tratamiento antibiótico tópico y oral.
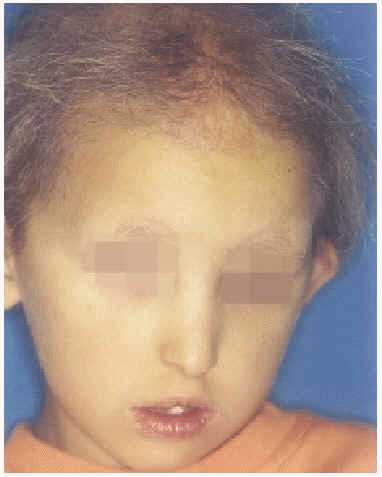
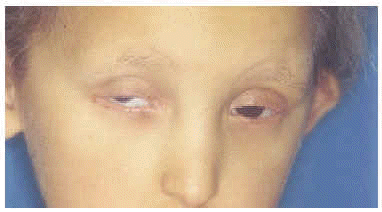
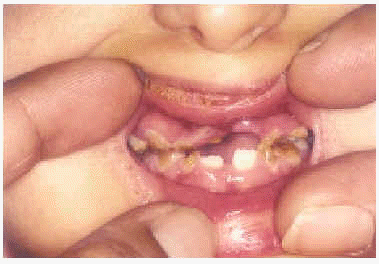
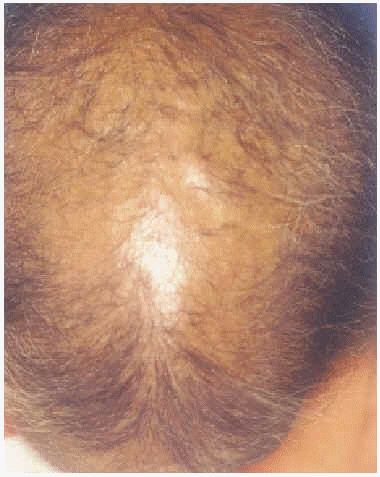
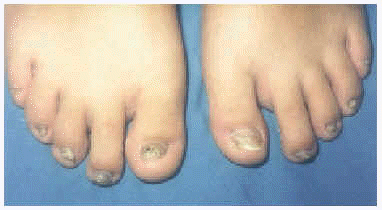
A la exploración física presentaba una facies con hipoplasia media, ptosis palpebral, deformidad en copa de pabellones auriculares (figs. 1 y 2) y dientes pequeños, algunos ausentes, con numerosas caries (fig. 3). En cuero cabelludo tenía pelo ralo y disperso, con atrofia e hiperpigmentación de la piel subyacente (fig. 4). En manos y pies se apreciaba distrofia ungueal de predominio distal (fig. 5). Por último, se observaban máculas hiperpigmentadas de aspecto residual en pliegues axilares e inguinales. La paciente presentaba un desarrollo psicomotor normal, precisando con frecuencia tratamiento antibiótico tópico y sistémico para las infecciones de pliegues y cuero cabelludo, y seguimiento para mantenimiento y restauración de las piezas dentarias.
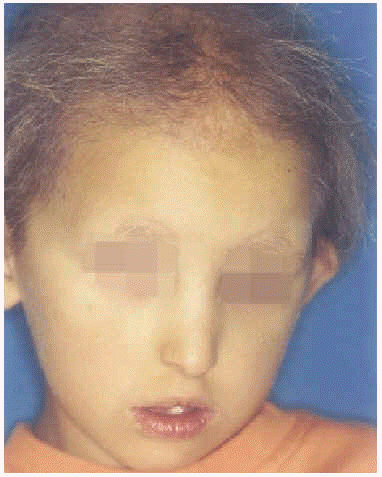
Fig. 1.--Facies hipoplásica con deformidad auricular y pelo escaso.
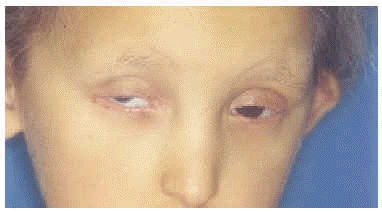
Fig. 2.--Ptosis palpebral, con anquilobléfaron intervenido.
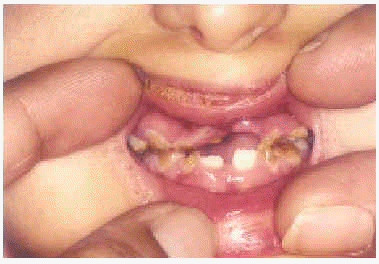
Fig. 3.--Hipodoncia y anodoncia con piezas cariadas.
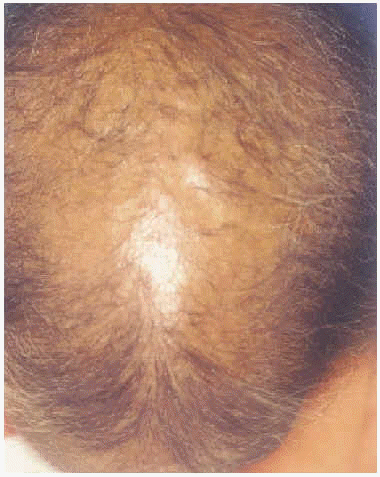
Fig. 4.--Pelo escaso y disperso con atrofia del cuero cabelludo.
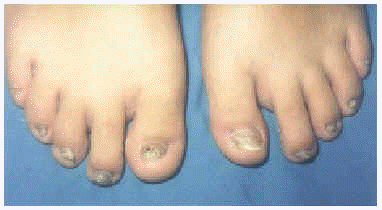
Fig. 5.--Hipoplasia ungueal distal.
DISCUSION
El síndrome AEC fue descrito por Hay y Wells4 en 1976 en 7 individuos pertenecientes a cuatro familias que presentaban un trastorno caracterizado por la presencia de anquilobléfaron, displasia ectodérmica y fisura labial y/o palatina, que se transmitía de forma autosómica dominante. La displasia ectodérmica consistía en afectación del pelo, que era basto y ralo en todo el cuerpo, de los dientes con hipodoncia o anodoncia, de las glándulas sudoríparas con leve hipohidrosis con intolerancia al calor y de la piel en forma de erosiones e infecciones predominantemente en cuero cabelludo. El anquilobléfaron o fusión de los párpados es el dato más característico, que aparece prácticamente en todos los pacientes. También es muy frecuente la fisura palatina, que aparece en el 90 % de los casos, mientras que la fisura labial es más inconstante en su aparición.
Además de todas las alteraciones mencionadas, pueden aparecer muchas otras, como por ejemplo atresia de los conductos lagrimales y coanas, deformidad auricular y otitis media con pérdida de audición secundaria y, de forma mucho menos frecuente, sindactilia, pezones supernumerarios y alteraciones genitales (fimosis, hipospadias, micropene o erosiones vaginales)4,5.
El diagnóstico diferencial del síndrome AEC se establece principalmente con otros dos trastornos que pueden presentar displasia ectodérmica y fisura labial/palatina: el síndrome EEC y el síndrome de Rapp-Hodgkin6. El síndrome EEC7 (ectrodactilia, displasia ectodérmica y fisura labial y/o palatina) presenta alteraciones muy similares a las descritas, pero además tiene de forma muy característica ectrodactilia, que es una anomalía de pies y/o manos en forma de hendidura longitudinal que les da un aspecto de «pinzas de cangrejo». El síndrome de Rapp-Hodgkin8 es mucho más difícil de distinguir del síndrome AEC, sobre todo cuando este último se presenta sin anquilobléfaron. Los pacientes con el síndrome de Rapp-Hodgkin suelen tener una facies muy característica, y en ellos son menos frecuentes e invalidantes las erosiones e infecciones en el cuero cabelludo5, aunque muchas veces existen características solapadas de ambos síndromes9. En tercer lugar, existen muchos otros cuadros parecidos al síndrome AEC, como el síndrome CLPED (displasia ectodérmica y fisura labial/palatina de herencia autosómica recesiva), el síndrome CHAND10 (que se diferencia del síndrome AEC únicamente por presentar de forma característica pelo rizado y retorcido), el síndrome de Bowen-Armstrong11 (que cursa con retraso mental e hiperpigmentación cutánea moteada), y otros.
En cuanto al trastorno genético responsable del síndrome AEC, parece que el llamado p63 podría estar implicado. El p63 es un factor de transcripción homólogo del p53 que se expresa en las superficies ectodérmicas12 y que se encuentra mutado en los síndromes AEC y EEC.13,14. En el síndrome AEC las mutaciones consisten en sustituciones de aminoácidos que afectan a la interacción entre proteínas. La mutación afecta a distintos dominios del ADN en los síndromes AEC y EEC, y aunque no se han demostrado hasta ahora mutaciones en p63 en el síndrome de Rapp-Hodgkin, se mantiene la hipótesis de que hay otros genes cercanos implicados en la patogenia de estas enfermedades3.



