


El síndrome de Sturge Weber es un trastorno neurocutáneo congénito, esporádico causado por una mutación somática activadora en el gen GNAQ, con una incidencia de uno de cada 20,000-50,000 nacidos. Se caracteriza por la presencia de una mancha en vino de Oporto facial, angiomatosis leptomeníngea y glaucoma. La manifestación neurológica más común son las convulsiones, que suelen comenzar en los primeros meses de vida. El glaucoma puede estar presente desde el nacimiento o desarrollarse posteriormente. Los estudios de neuroimagen permiten visualizar la angiomatosis leptomeníngea, ayudando al diagnóstico del síndrome de Sturge Weber. El tratamiento estándar incluye láser para la mancha en vino de Oporto facial, anticonvulsivantes y tratamiento médico o quirúrgico del glaucoma. El pronóstico de la enfermedad dependerá de la extensión de la malformación leptomeníngea y del grado de afectación ocular.
Sturge-Weber syndrome is a sporadic congenital neurocutaneous disorder caused by a somatic activating mutation in GNAQ; it affects 1 in every 20,000 to 50,000 newborns. It is characterized by a facial Port-wine stain, leptomeningeal angiomatosis, and glaucoma. Seizures are the most common neurological manifestation and typically present in the first months of life. Glaucoma may be present at birth or develop later. Neuroimaging studies show leptomeningeal angiomatosis, supporting diagnosis. Standard treatment for Sturge-Weber syndrome includes laser treatment for the Port-wine stain, anticonvulsants, and medical or surgical treatment for the glaucoma. Prognosis depends on the extent of leptomeningeal involvement and the severity of the glaucoma.

El síndrome de Sturge Weber (SSW) es un trastorno neurocutáneo, que en su forma completa asocia una malformación capilar (mancha en vino de Oporto [MVO]) facial, glaucoma y angioma leptomeníngeo. Su incidencia se estima en uno de cada 20.000-50.000 nacidos vivos1. Hay quien emplea el nombre de Sturge-Weber para denominar formas incompletas en las que solo existen 2 de las manifestaciones. En este sentido hay quien clasifica las «angiomatosis» encefalofaciales según la escala de Roach en 3 tipos: tipo i con MVO facial y angiomatosis leptomeníngea, con o sin glaucoma asociado que correspondería SSW clásico; tipo ii, que es el más frecuente, con MVO facial sin afectación leptomeníngea, con presencia o no de glaucoma; y tipo iii, que es la forma menos frecuente, con la presencia únicamente de angiomatosis leptomeníngea2.
EmbriologíaLa malformación capilar (MVO) y la angiomatosis leptomeníngea podrían ser el resultado de un fallo en la regresión del plexo venoso cefálico primitivo. En los estadios iniciales, el sistema venoso primitivo se divide en una porción externa, que abastece y drena la piel de la cara y del cuero cabelludo, una porción media, que irriga las meninges, y una porción profunda, que abastece y drena el cerebro3. Se ha sugerido que en este estadio la proximidad embriológica del ectodermo destinado a formar el área superior de la piel de la cara con la parte del tubo neural que formara el área parieto-occipital del cerebro, podría explicar la asociación de la MVO facial con el angioma leptomeníngeo4,5.
Genética y patogénesisDurante años se postuló que la causa del SSW podría ser una mutación somática, y que ello explicaría su carácter esporádico y la distribución parcheada de los vasos sanguíneos anormales4,6.
En el año 2013 Shirley et al.7 detectaron una mutación somática activadora en el gen GNAQ, concretamente la sustitución de un solo nucleótido (c.548G→A, p.Arg183Gln) en 23 muestras de tejido afectado de 26 pacientes con SSW (88%), y en 12 de 13 pacientes con MVO no sindrómicas (92%). La prevalencia de la mutación en los tejidos afectados estaba entre el 1-18% confirmando su carácter de mosaico7. Otros autores han confirmado que el 80% de los pacientes con SWS tiene esta mutación somática en el tejido cerebral8.
La extensión de la MVO, la presencia de afectación leptomeníngea y ocular dependerá del momento en que se produzca dicha mutación y la célula afecta8.
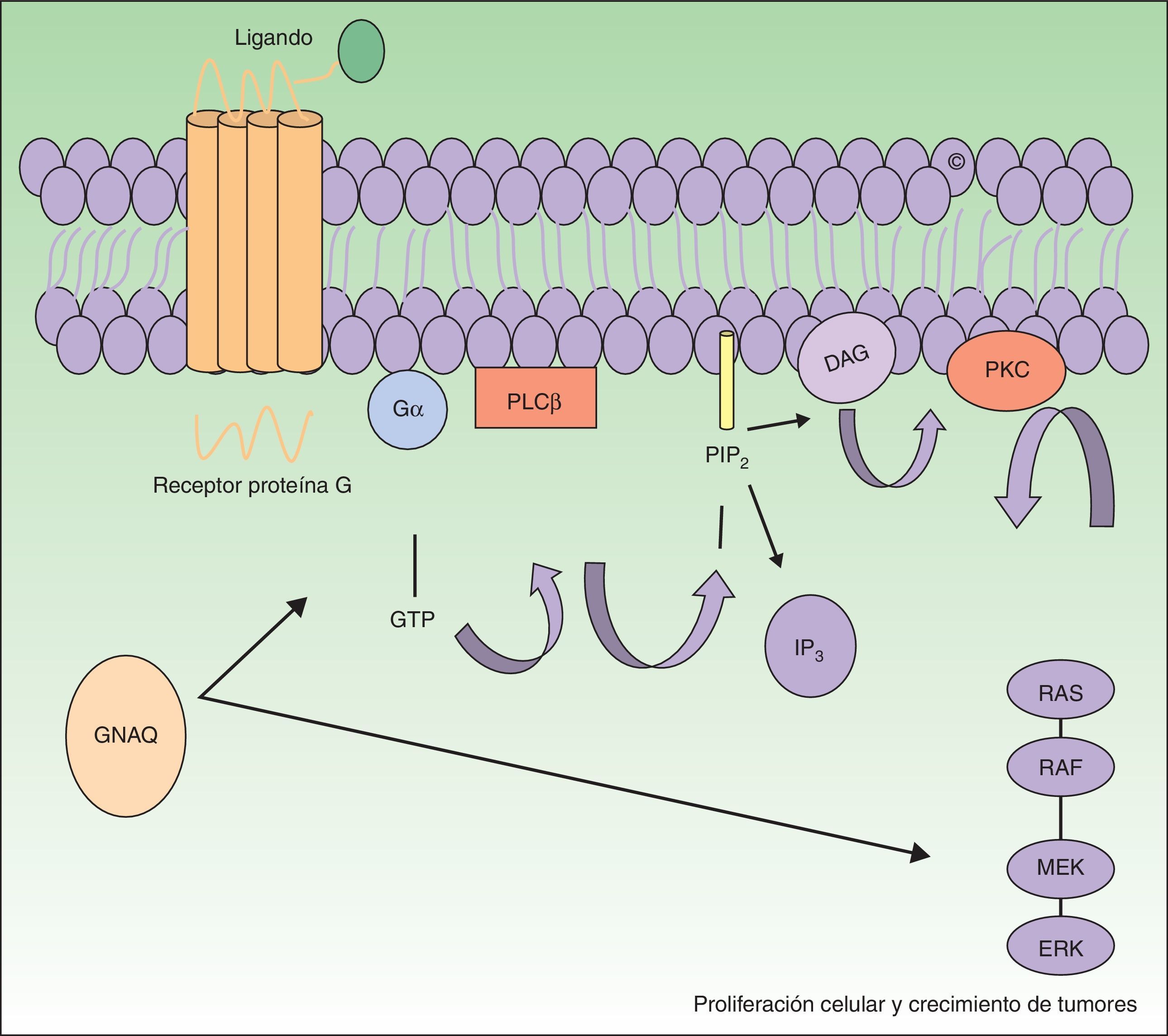
El gen GNAQ se halla en el cromosoma 9 y se compone de 7 exones que abarcan una región de 310,993 nucleótidos. Es uno de los 4 miembros pertenecientes a la familia Gq-alfa. Las proteínas Gq son una clase de proteína G que participan como moduladores o transductores en los diferentes sistemas de señalización transmembrana. Mutaciones somáticas en GNAQ, diferentes a las detectadas en malformaciones capilares, han sido descritas en neoplasias melanocíticas, en melanoma uveal, hemangiomas congénitos (RICH y NICH) y facomatosis pigmentovascularis9,10. En esta última entidad se han detectado mutaciones diversas en mosaico en GNAQ en GNA11, y al parecer en los casos en los que se ha estudiado se ha podido detectar la misma mutación en el componente pigmentario y en el vascular11. Estas mutaciones aumentan la proliferación e inhiben la apoptosis debido al incremento de señalización a través de las vías efectoras RAS (fig. 1).

El GNAQ está implicado en el crecimiento celular a través de la transmisión de señales por medio de los receptores de la membrana celular vía MAP cinasas. Por tanto, una mutación activadora aumentaría la señalización por esta vía, lo que puede conducir a las malformaciones capilares observadas en el síndrome de Sturge Weber.
DAG: 1,2 diacilglicerol; Gα: proteína G alfa; IP3: 1,4,5 trifosfato; PIP2: fosfatidilinositol 4,5-bifosfato; PKC: proteína C cinasa; PLCβ: fosforilasa C.
La malformación capilar o venular, también denominada mancha en vino de Oporto o nevus flammeus, es la característica cutánea fundamental en el SSW12. Se trata de una mancha que ya está presente en el momento del nacimiento, de tamaño variable, normalmente lateralizada, aunque puede ser bilateral, y de color que oscila de rosa pálido a purpúrico. La MVO se puede confundir con una mancha salmón (nevus simplex). La mancha salmón es una malformación capilar que se presenta como una mácula color rosa mal delimitada, localizada habitualmente en la zona central de la frente, philtrum, párpados superiores, vertex y nuca13. La localización lateralizada de la MVO, su color más intenso y sus límites bien delimitados permite en la mayoría de los casos diferenciarla de la mancha salmón.
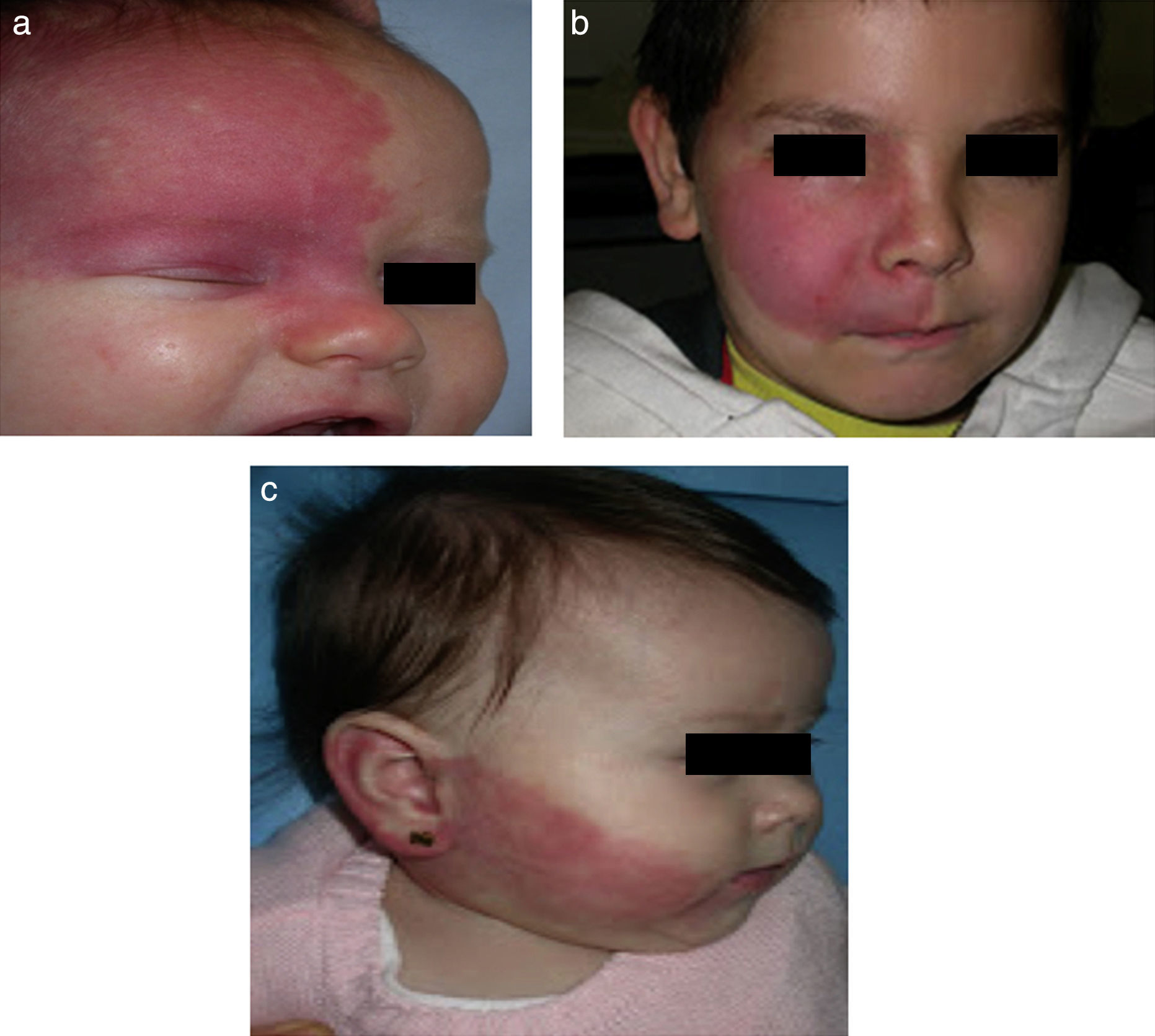
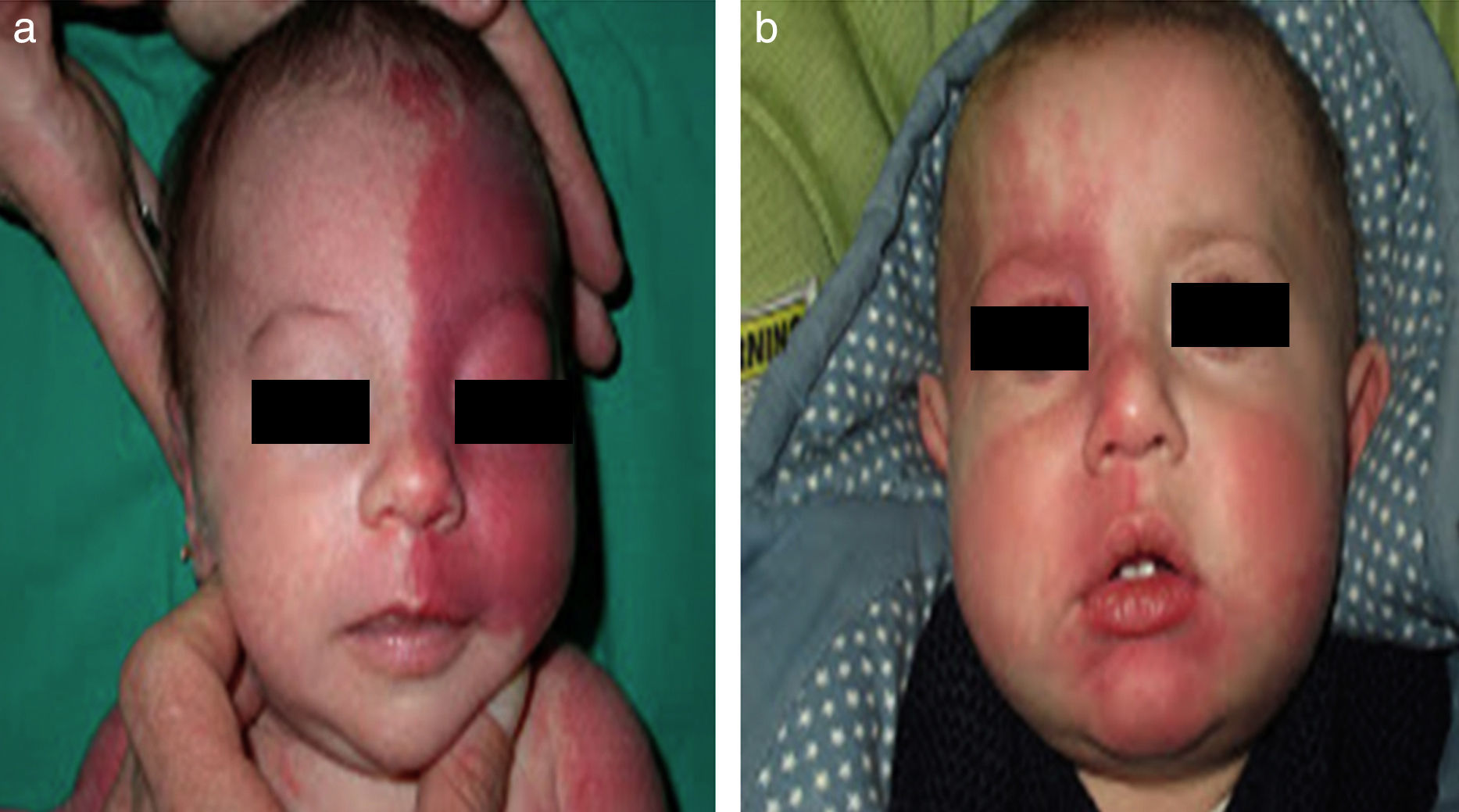
El riesgo de que una malformación capilar (MVO) facial asocie afectación leptomeníngea u ocular depende de la extensión de la MVO y de su localización. Así, las malformaciones que afectan a la zona frontal tienen mayor riesgo que las que afectan a la parte inferior de la cara14. Hasta la fecha los estudios de riesgo de asociación lo han estratificado considerando que las MVO faciales tienen una distribución que remeda el área de inervación sensitiva de las 3 ramas del trigémino: la rama frontal (V1), la maxilar (V2), y la mandibular (V3)15–18 (figs. 2a-c). Todos los estudios coinciden en señalar que las MVO que afectan a la V1, párpado superior, o las que son bilaterales y extensas son las que tiene mayor riesgo de asociación (figs. 3a y b). Así, la incidencia de afectación leptomeníngea o glaucoma en los pacientes con malformaciones capilares que afectan V1 es aproximadamente del 8-15%, mientras que si la afectación es bilateral o afecta a varios dermatomas es del 28%17. El riesgo de que las MVO que solo afectan V2 o V3 asocien afectación leptomeníngea o glaucoma es prácticamente nula, según la mayoría de los autores17,18. Sin embargo, en algunos estudios la afectación de V2 se considera que tiene un riesgo del 2%17,18. Esta discrepancia puede ser debida a que hay autores que consideran el párpado inferior como V2 y hay otros que lo consideran como aparte de V117,18.
 rama frontal (V1); b) rama maxilar (V2); c) rama mandibular (V3).")
 Mancha en vino de Oporto con afectación extensa; b) mancha en vino de Oporto con afectación bilateral.")
Esta distribución de las MVO según las ramas de inervación sensitiva del trigémino ha sido objeto de controversia. Waelchli et al.13, después de revisar la distribución de las MVO faciales en 192 niños, llegaron a la conclusión de que no siguen las áreas sensitivas del trigémino, sino que siguen 8 áreas más o menos repetitivas de distribución. Según estos autores la zona de la frente, que es la que está comprendida desde la línea media de la frente a una línea que une el canto externo del ojo hasta la parte superior de la oreja, y que incluye el párpado superior, es la única que tendría riesgo de asociación. En este estudio todos los pacientes con alteraciones en la resonancia magnética (RM), convulsiones o glaucoma tenían afectación de esta área frontal. Hay que señalar, sin embargo, que en este estudio retrospectivo solo se realizó RM en 4 de los pacientes con MVO fuera de esta área. El área frontal remeda la distribución de la prominencia frontonasal, y la piel en el área de la vesícula óptica, que derivan de la placoda frontal. Las placodas desarrollan su propia vasculatura, por lo que estos autores postulan que las MVO de la cara remedan la vasculatura embriológica y no siguen la distribución de inervación sensitiva de la cara. La placoda frontal desarrolla su vasculatura a partir del prosencefalo y el mesencéfalo anterior, estructuras que darán lugar al cerebro y a la vesícula óptica, por lo que explicaría que las MVO de esta zona derivada de la placoda frontal son las que tengan riesgo de SSW13,19.
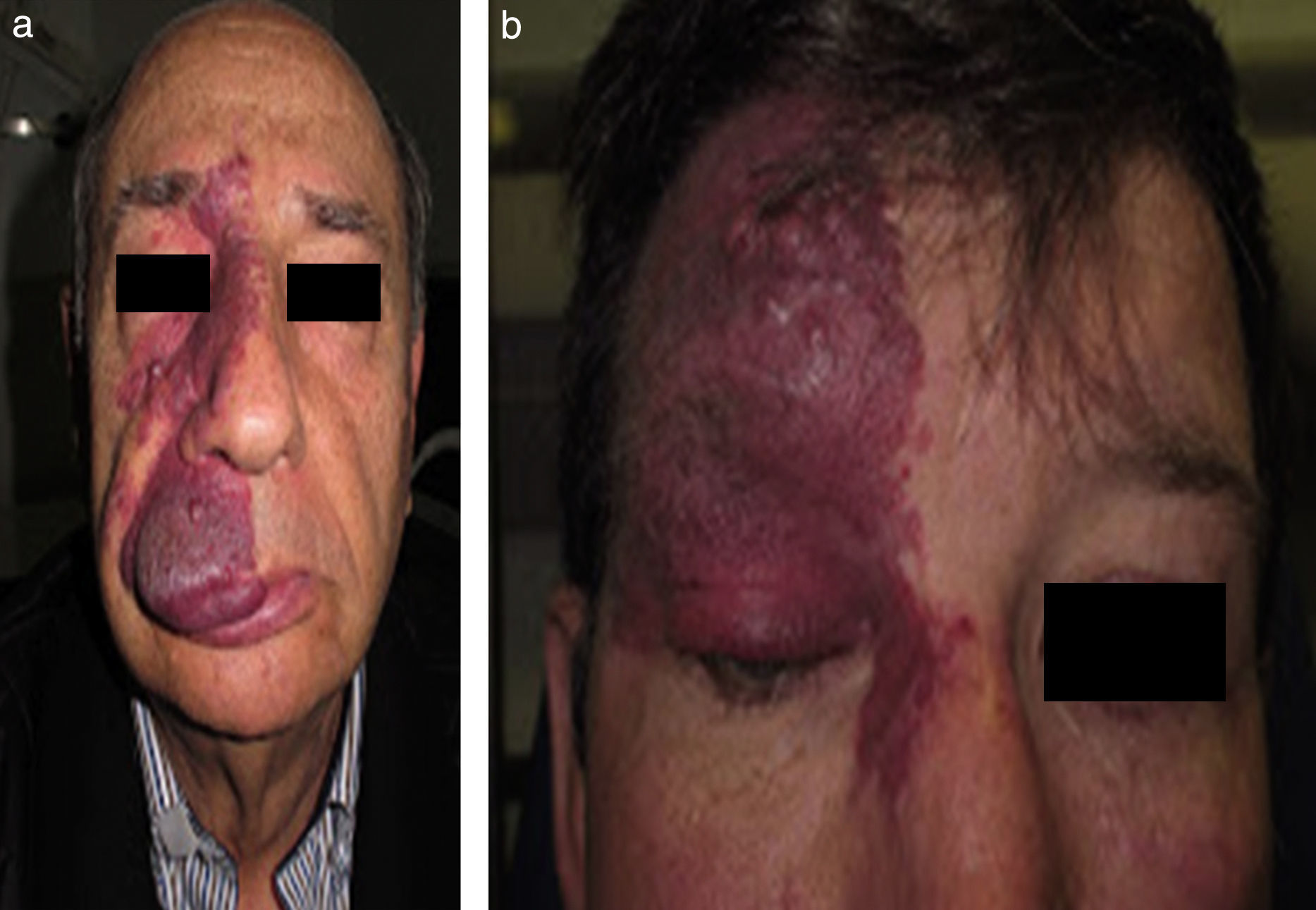
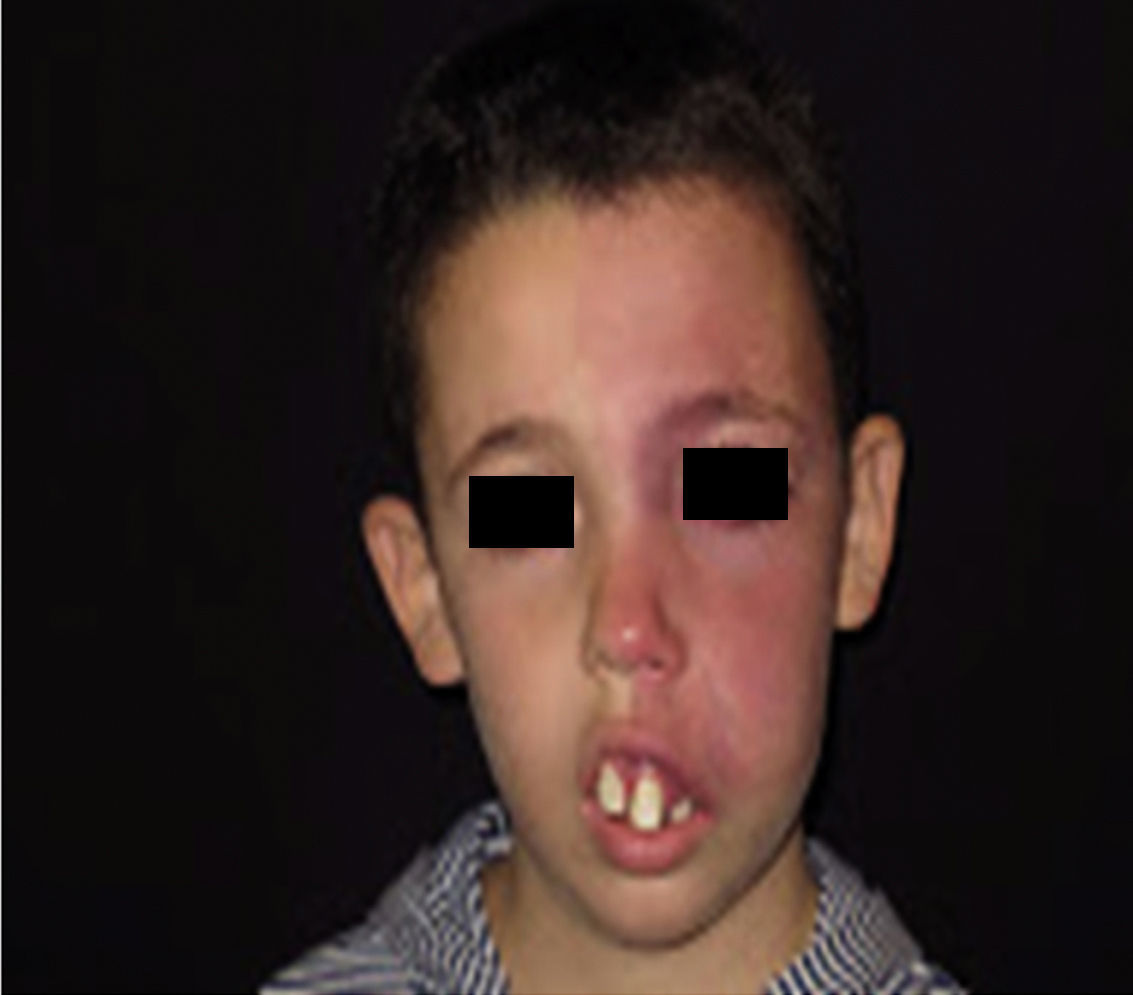
La MVO facial puede desarrollar en un 60% hipertrofia de tejidos blandos, en un 13,8% hipertrofia ósea y en un 43,8% la formación de nódulos proliferativos o de ectasia progresiva (figs. 4a y b). La edad media del inicio de estos cambios es a los 9 años y el segmento maxilar (V2) es el que con mayor frecuencia presenta hipertrofia20,21. Debido al crecimiento excesivo del maxilar y de la mandíbula se observa una mala oclusión y una mayor exposición dental, conduciendo a una deformidad facial significativa22 (fig. 5).
 Mancha en vino de Oporto con hipertrofia de tejidos blandos; b) mancha en vino de Oporto con formación de nódulos proliferativos.")

Una de las manifestaciones del SSW es la malformación leptomeníngea capilar-venosa. La angiomatosis intracraneal suele ser ipsilateral a la MVO, si bien puede ser bilateral. Generalmente afecta el lóbulo occipital, occipito-parietal y en ocasiones a todo el hemisferio1.
Histológicamente la malformación vascular cerebral consiste en estructuras vasculares tortuosas y anormales en las leptomeninges engrosadas. También se observan alteraciones en las venas profundas de drenaje que se encuentran anormalmente dilatadas. El tejido cerebral subyacente puede ser atrófico y mostrar una pérdida neuronal, astrocitosis, disgenesia cortical y las calcificaciones en distribución perivascular o en el córtex cerebral. Se cree que se trata de calcificaciones distróficas secundarias a la hipoxia. Los vasos leptomeníngeos de pacientes con SSW muestran tanto una sobreexpresión de fibronectina y factor de crecimiento endotelial vascular (VEFG), como un incremento de la proliferación endotelial y apoptosis4,23.
Las principales manifestaciones clínicas de la angiomatosis leptomeníngea son convulsiones (75-90%), hemiparesia lentamente progresiva (25-60%), cefaleas vasculares de tipo migrañoso (30-45%), retraso del desarrollo neuropsicológico (50-60%), episodios similares a eventos cerebrovasculares, con hemiplejías agudas transitorias, defectos del campo visual y problemas de conducta1,3. Las convulsiones presentes en los pacientes con SSW son el resultado de la irritabilidad cortical causada por la malformación vascular cerebral, a través de mecanismos de hipoxia, isquemia y gliosis24. Las convulsiones suelen ser focales o parciales complejas con generalización secundaria23. En el 75% de los pacientes con afectación cerebral las convulsiones se presentan durante el primer año de vida y en el 90% dentro de los primeros 2 años25. Un episodio febril puede precipitar el comienzo de las convulsiones en el SSW. Los pacientes sin MVO, pero con angiomatosis leptomeníngea, pueden presentar convulsiones durante la infancia o incluso en la edad adulta14. Los pacientes que inician las crisis convulsivas antes de los 2 años de edad tienen convulsiones más difíciles de controlar y un mayor riesgo de retraso mental26,27. El deterioro neurológico progresivo de los niños con SSW es de tipo isquémico por la alteración de la perfusión cerebral, asociada a una demanda metabólica elevada por la actividad convulsiva prolongada4.
Manifestaciones ocularesLa malformación vascular del ojo consta de vasos venosos dilatados y tortuosos, que puede afectar la conjuntiva, la epiesclera, la retina y/o la coroides, terminando en atrofia óptica y ceguera23.
El glaucoma es una de las manifestaciones oculares más frecuentes observadas en pacientes con SSW, afectando alrededor del 30-70% de pacientes25,28,29. Suele ser unilateral e ipsilateral a la MVO, pero no siempre es así1. El glaucoma en el SSW se considera que puede ser secundario bien al aumento de la presión venosa epiescleral, mecanismo fisiopatológico apoyado por la presencia de sangre dentro del canal de Schlemm, bien por anomalías de la cámara anterior que interfieren con el drenaje normal del humor acuoso, con el consiguiente aumento de la resistencia a la salida23,30. El glaucoma puede ser congénito o tardío. Las formas tempranas son, en el 60% de los casos, secundarias a anomalías del ángulo de la cámara anterior, mientras que la causa de glaucoma en el 40% de los jóvenes y adultos jóvenes son la elevada presión venosa epiescleral30.
El hemangioma coroideo está presente en el 40-50% de los pacientes con SSW, bien sea de forma circunscrita y/o difusa. En ocasiones la lesión vascular de la coroides adopta un carácter patognomónico, como un resplandor rojizo, a la que se le ha aplicado el término descriptivo fondo de «tomate cátsup»24. La coroides normalmente permanece sin cambios durante toda la infancia; sin embargo, en la adolescencia o la edad adulta, en ocasiones se vuelve notablemente engrosada30.
Se han descrito otras anomalías oculares menos frecuentes como son: heterocromía del iris, desprendimiento de retina, estrabismo, hemianopsia homónima, facomatosis pigmento vascular, neovascularización del iris y la coroides y luxación del cristalino31,32.
Manifestaciones endocrinasLos pacientes con SSW pueden tener una disfunción hipotálamo-hipofisaria. con déficit de hormona del crecimiento e hipotiroidismo central33–35.
Exploraciones complementariasRadiografía de cráneoEn las radiografías simples, aunque no es un estudio de elección, pueden observarse en ocasiones las clásicas calcificaciones corticales giriformes también llamadas en vía de ferrocarril, que afecta a la capa íntima de las arterias meníngeas. Se encuentran adyacentes al angioma leptomeníngeo, principalmente en la región parietal y occipital26,36. Las calcificaciones suelen afectar a niños mayores de 2 años, por lo que son un hallazgo tardío37.
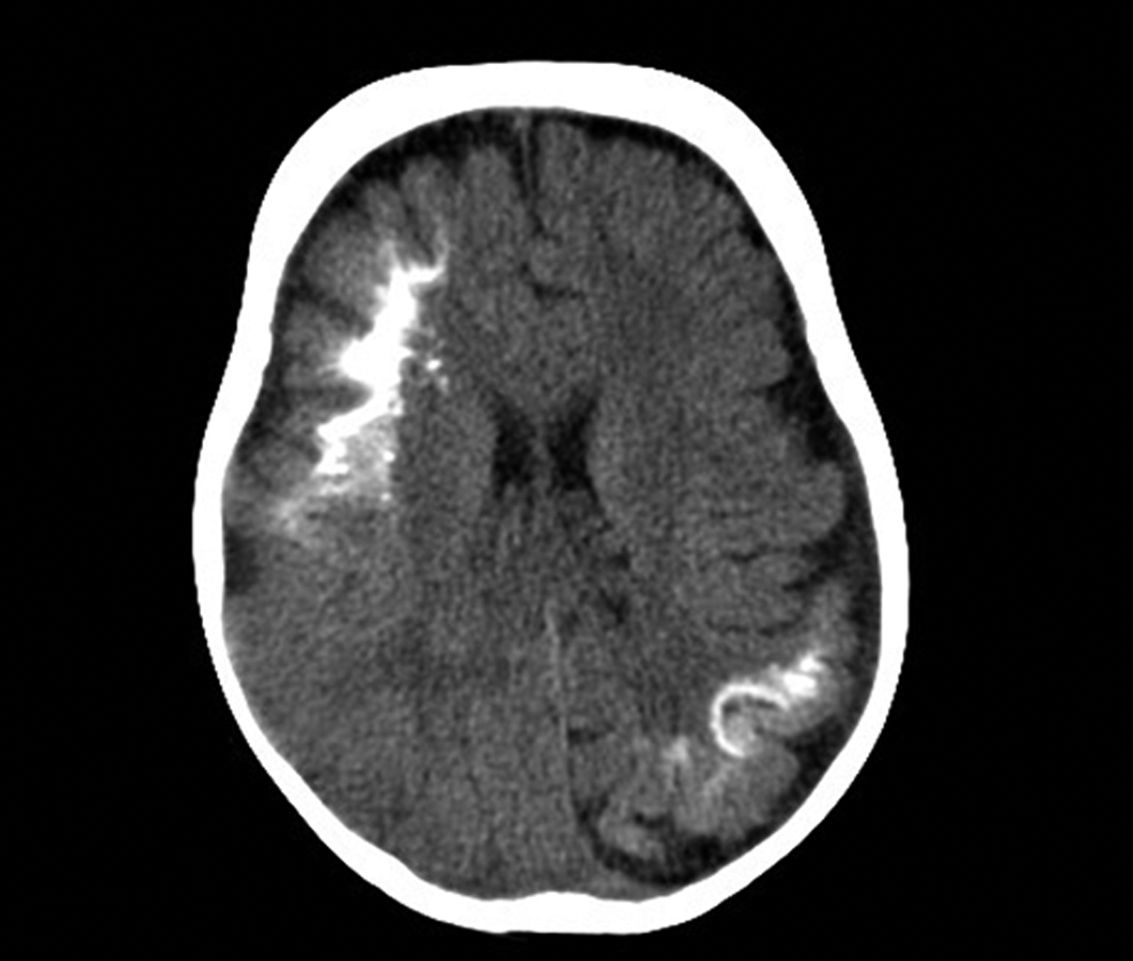
Tomografía axial computarizadaLa tomografía cerebral es una técnica que se utiliza frecuentemente en urgencias, para la valoración de un niño que comienza con hemiparesia o convulsiones. Los hallazgos característicos son la presencia de pérdida de volumen parenquimatoso, agrandamiento del ventrículo y el agrandamiento del plexo coroideo. A partir del año de edad la tomografía axial computarizada puede detectar calcificaciones (fig. 6), que normalmente no serían visibles en una radiografía simple, y es más sensible en la detección de calcificaciones que la RM14,37.
Resonancia magnética cerebral
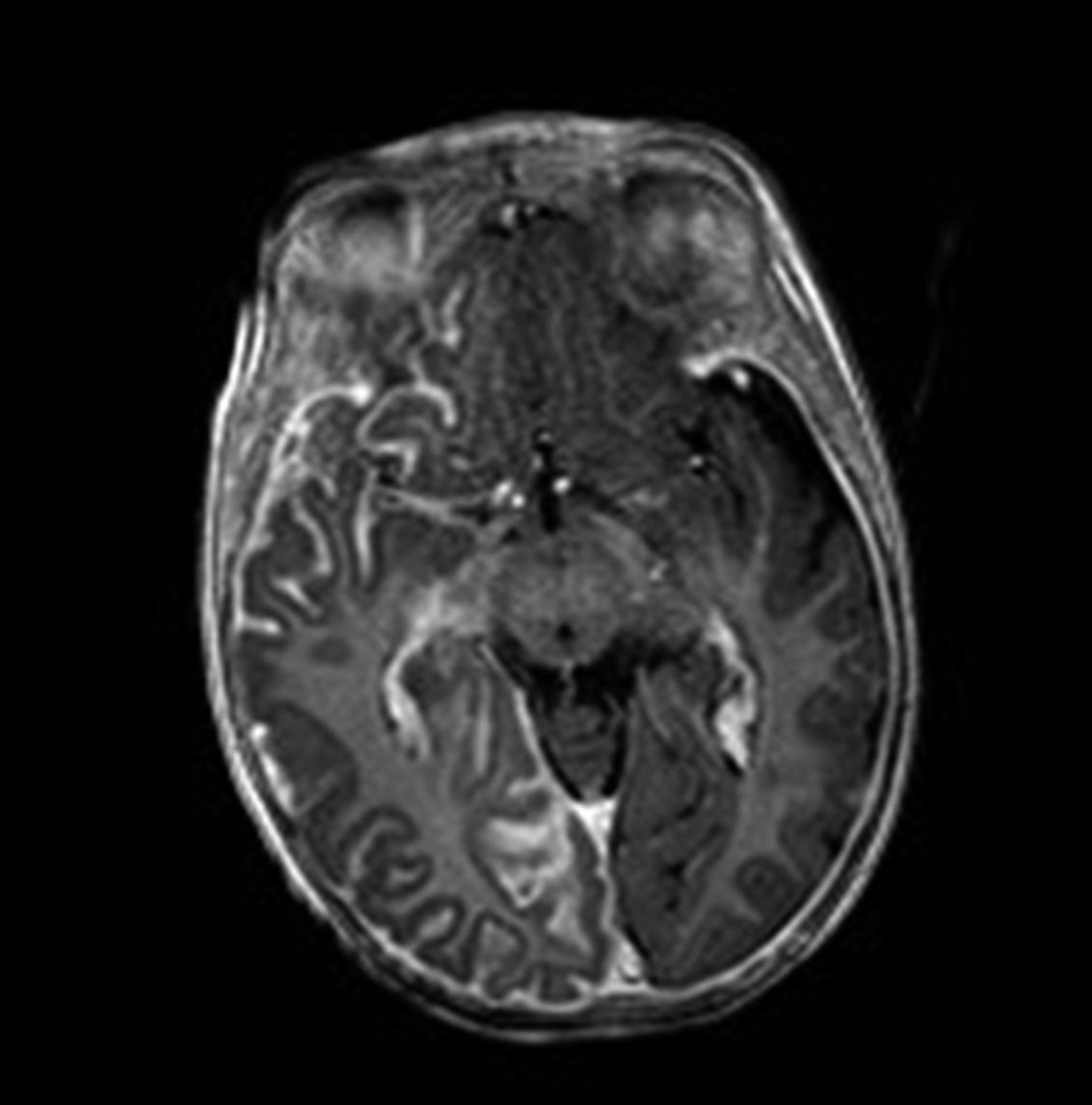
La RM cerebral con gadolinio es la técnica de imagen de elección para el diagnóstico del SSW28. Con esta técnica es posible visualizar la malformación vascular leptomeníngea que confirma el diagnóstico del SSW (fig. 7). Las calcificaciones en la RM también se pueden detectar en secuencias T2 como imágenes de hiposeñal cortical o yuxtacortical. La resonancia permite además demostrar un drenaje venoso anómalo, una reducción del volumen cerebral, un agrandamiento del plexo coroideo ipsilateral, prominencia de las venas subependimarias y medulares, pérdida de volumen del hemisferio cerebral efecto y una mielinización acelerada subyacente al angioma leptomeníngeo14,24,37. Estos cambios son más evidentes después del año de edad38.
Electroencefalograma
El EEG característico de pacientes con SSW es asimétrico, con una reducción del voltaje y descargas focales en el hemisferio cerebral afectado. El EEG también es bastante útil para distinguir migrañas y eventos cerebrovasculares de convulsiones como causas de los eventos paroxísticos agudos1.
El EEG en pacientes con SSW parece evolucionar con el tiempo, haciéndose cada vez más anormal, con actividad epileptiforme más frecuente39.
Imágenes de perfusiónDurante los primeros meses de vida la malformación vascular tiende a estar hiperperfundida. El patrón pasa a hipoperfusión al final del primer año, incluso en pacientes sin convulsiones24,37, aunque este está acelerado por las crisis y la progresiva isquemia, hipoxia y privación de glucosa en el parénquima subyacente a la malformación, que son responsables del deterioro neurológico progresivo en los pacientes con SSW.
AngiografíaLa angiografía no se realiza de forma rutinaria, resulta útil en casos atípicos para evaluar otras anomalías vasculares asociadas (oclusiones venosas, lesiones trombóticas o malformaciones arteriovenosas) o en la identificación de los grandes vasos diploicos, evitándose así sangrados en el caso de que sea necesaria una craneotomía24.
DiagnósticoEl diagnóstico de SSW debe sospecharse en pacientes con MVO en la zona de la frente (fig. 8). Con esta sospecha debe realizarse una revisión oftalmológica y una RM con contraste, para realizar un diagnóstico temprano y reducir las complicaciones oftalmológicas y cerebrales13. Los estudios de imagen tienen un papel importante en el diagnóstico, la detección y el seguimiento de los pacientes con SSW.

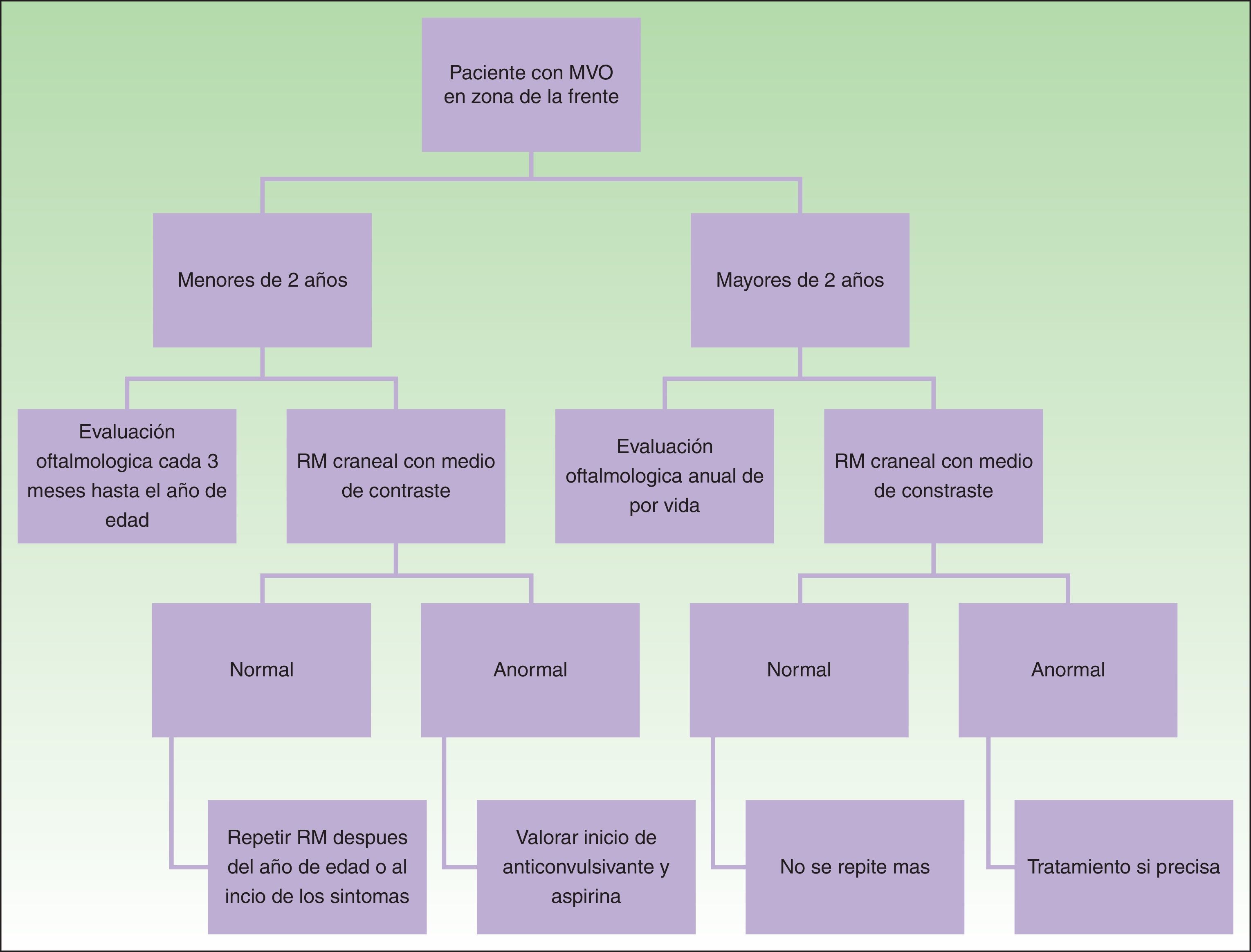
La necesidad de realizar una RM craneal en todos los pacientes con MVO en una localización de riesgo es controvertida, puesto que existe un pequeño porcentaje de falsos negativos, y por otra parte no todos los pacientes con afectación leptomeníngea van a presentar convulsiones (10%)24. Dado que el porcentaje de falsos negativos es pequeño, y que el detectar angiomatosis leptomeníngea puede ayudar a dar información a los padres sobre cómo detectar convulsiones o cómo actuar en caso de una convulsión, nosotros preferimos realizarla aun en pacientes asintomáticos. La angiomatosis leptomeníngea puede ser difícil de visualizar antes de los 3 meses de edad, por lo que se recomienda obtenerla entre los 3-6 meses de edad14.
En caso de que el paciente se presente con una MVO en localización de riesgo después de los 2 años de edad, es más discutible la necesidad de completar el estudio con RM en un paciente asintomático porque es poco probable que tenga convulsiones de difícil control si no las ha tenido previamente1.
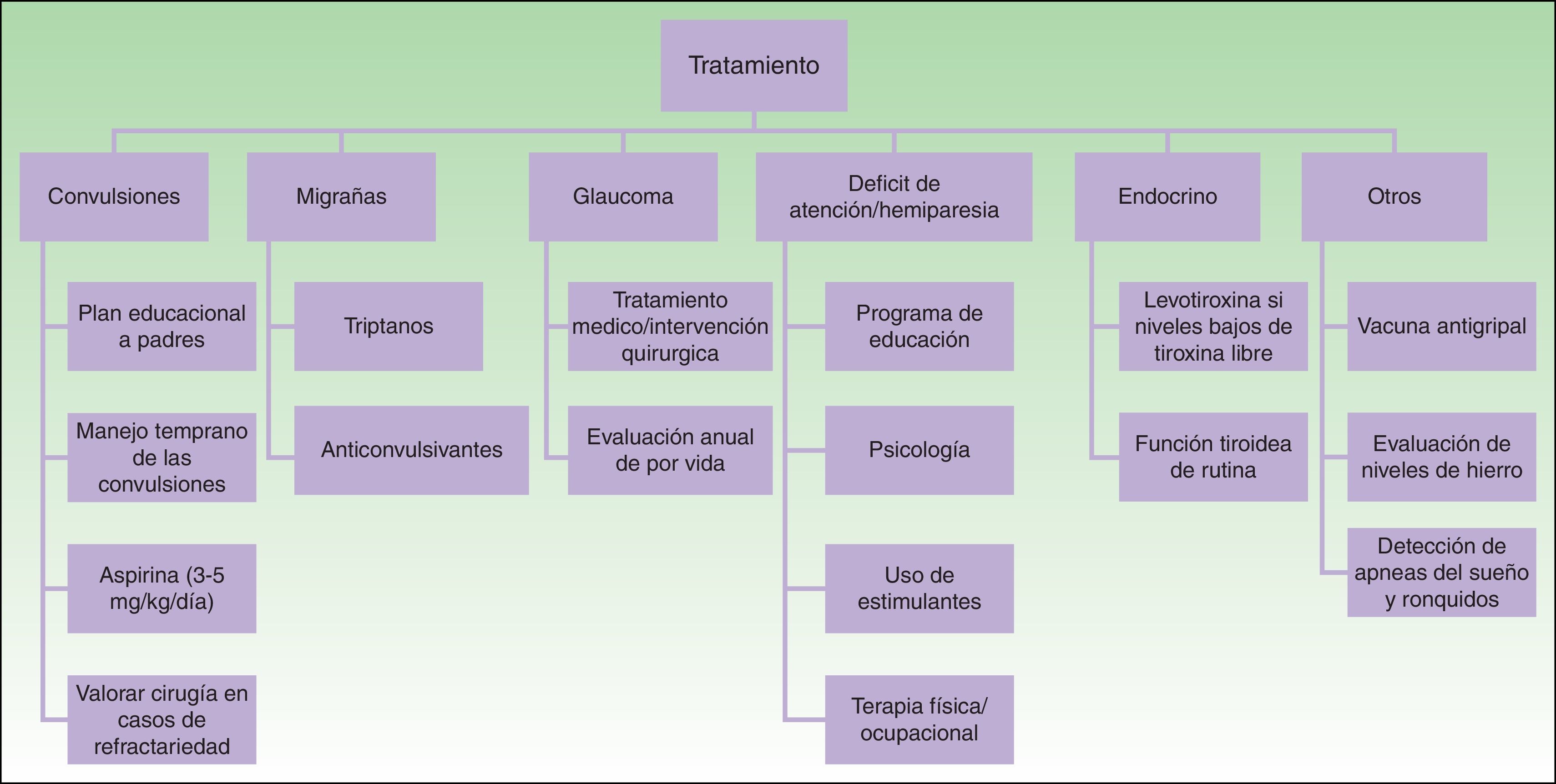
TratamientoEl tratamiento médico en pacientes con SSW incluye anticonvulsivantes, tratamiento sintomático y profiláctico para el dolor de cabeza, tratamiento del glaucoma para reducir la presión intraocular y la terapia con láser para la MVO24 (fig. 9).

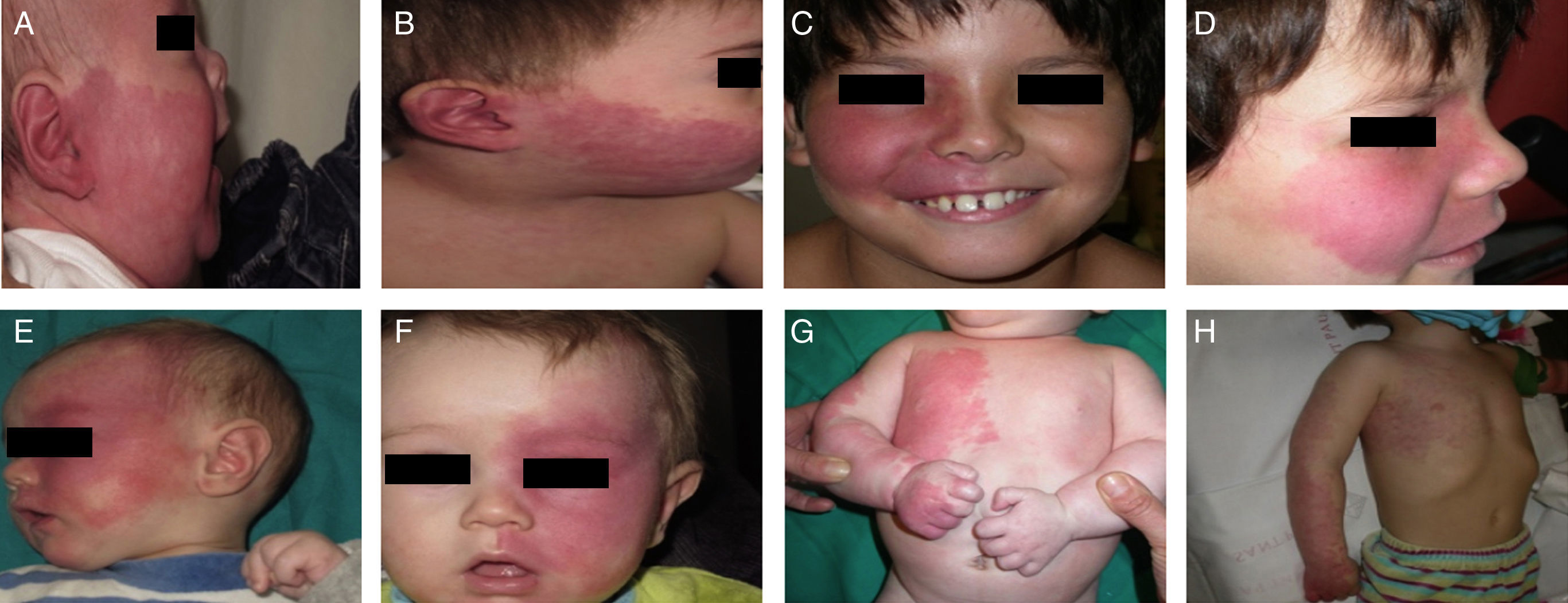
El tratamiento con láser colorante pulsado (PDL) es el tratamiento de elección para la MVO facial40. En general son necesarias entre 7-15 sesiones para conseguir el aclaramiento de las lesiones, y raramente se consigue una desaparición completa41,42. El grado de respuesta depende del color inicial de la lesión y de la zona en la que esté situada. Por lo general, las MVO de la región frontal responden mejor que las de la zona malar o el prolabio43. (fig. 10a,b,c,d,e,f,g,h). La mayoría de autores coinciden en que las respuestas suelen ser mejores si el tratamiento se realiza durante la infancia que en la edad adulta, si bien no existe evidencia sólida al respecto. En general se recomienda iniciar el tratamiento tan pronto como sea posible43–46. Aun con todo, ante una MVO determinada es difícil predecir a priori el grado de respuesta que se va a obtener. La resistencia terapéutica es multifactorial, pero se cree que en parte es debido a la regeneración y revascularización de los vasos fotocoagulados. Ensayos clínicos han demostrado la obtención de mejores resultados y una reducción del número de sesiones necesarias con la asociación de rapamicina, tanto en su forma tópica como oral, inmediatamente después del tratamiento con PDL. Aun con todo el uso de rapamicina tópica para esta indicación todavía no constituye una práctica común, y el uso de rapamicina oral no parece justificada para esta indicación47,48.
 zona mandibular y zona malar color rosa intenso, difícil de tratar; e y f) zona frontal y lateral de la cara con mejor respuesta que la zona central; g y h) zona de extremidades y tronco, peor respuesta en las zonas acrales. Véase el buen aclaramiento de la zona pectoral y del brazo.")
Antes y después de 7 sesiones con láser y diferencia de respuesta según la localización; a,b,c,d) zona mandibular y zona malar color rosa intenso, difícil de tratar; e y f) zona frontal y lateral de la cara con mejor respuesta que la zona central; g y h) zona de extremidades y tronco, peor respuesta en las zonas acrales. Véase el buen aclaramiento de la zona pectoral y del brazo.
La luz pulsada intensa, el láser de alejandrita, el láser secuencial PDL y el Ndyag también se han utilizado para el tratamiento de la MVO. A pesar de su buen funcionamiento, el tratamiento con PDL sigue siendo el tratamiento de elección49.
La clave para prevenir la progresión del daño neurológico en el SSW es el temprano reconocimiento y el manejo de las convulsiones, desarrollando un plan de actuación con los padres y los hospitales locales. Es importante que los padres sean instruidos sobre cómo reconocer una convulsión y cómo actuar en caso de que ocurra. Por otro lado, y dado que la fiebre o las enfermedades intercurrentes pueden precipitar las crisis convulsivas, es importante un buen control de la fiebre, asegurar una buena hidratación y control de la oxigenación1,23.
Otras medidas importantes en el control de las convulsiones de pacientes con SSW incluyen: la administración anual de la vacuna antigripal, el aumento de las dosis de anticonvulsivantes según el peso, descartar anemias ferropénicas, así como detección y tratamiento de apneas del sueño que puedan agravar la isquemia cerebral14.
Los anticonvulsivantes siguen siendo la piedra angular del tratamiento de las convulsiones. Los anticonvulsivantes comúnmente utilizados en lactantes incluyen carbamazepina, oxcarbazepina, levetiracetam y fenobarbital50. La carbamazepina y la oxcarbazepina resultan tener un mejor control de las convulsiones en pacientes con SSW51.
Dado que las convulsiones determinan el deterioro cognitivo de los pacientes con SSW se ha planteado la utilidad de realizar tratamiento preventivo de las convulsiones con antiepilépticos, especialmente en pacientes con afectación leptomeníngea extensa50. En este sentido el uso «profiláctico» de fenobarbital antes de la primera convulsión se asocia con un menor riesgo de deterioro cognitivo en pacientes que lo reciben antes de su primera convulsión, versus los que lo recibieron después de su primera crisis52. Esta es una práctica que no se puede generalizar por la toxicidad del fenobarbital, pero a considerar en pacientes con afectación cerebral bilateral extensa50.
Otra medida profiláctica del deterioro neurológico que se ha planteado es la Aspirina® a dosis antiagregantes. Los pacientes con SSW a menudo tienen episodios de isquemia vascular, secundaria a la estasis venosa y a fenómenos trombóticos. Algunos estudios demuestran que dosis bajas de ácido acetilsalicílico a 3-5mg/kg/día disminuyen la frecuencia y la gravedad de los eventos cerebrovasculares y las crisis convulsivas53,54. Los efectos adversos que se pueden observar con el uso de Aspirina® son un incremento de hematomas, hemorragias nasales y sangrado de encías55.
Las convulsiones llegan a ser intratables en el 30-50% de pacientes con SSW. En estos casos estaría indicada la cirugía temprana, bien mediante lesionectomía, bien por callosotomía y hemisferestomía1,23,50. En un estudio de 20 pacientes con SSW se demostró que la hemisferestomía tiene un 90% de efectividad en la eliminación de las convulsiones, siendo más eficaz que la resección focal56. Algunos autores han descrito el uso de triptanes y anticonvulsivantes para el tratamiento de los dolores de cabeza24.
Los pacientes con déficit cognitivos y problemas de atención con hiperactividad pueden beneficiarse de servicios especializados en educación, psicología del comportamiento, así como el uso de estimulantes (metilfenidato o dextroanfetamina)57. En pacientes con hemiparesia es importante iniciar terapia física y ocupacional cuanto antes. Así mismo, se ha de realizar una evaluación neuropsicológica en niños más mayores50,58.
El principal objetivo en el tratamiento del glaucoma en pacientes con SSW es el control de la presión intraocular, para prevenir el daño del nervio óptico30. El tratamiento del glaucoma de aparición temprana o con anomalías del ángulo camerular, por lo general es quirúrgico, ya sea goniotomía o trabeculectomía. La tasa de éxito de la cirugía del ángulo es generalmente menor que en el glaucoma congénito primario, y a menudo requiere cirugía adicional, como la trabeculectomía o mediante la colocación de un dispositivo de drenaje. La principal complicación de la intervención quirúrgica es la hemorragia retiniana, por la disminución demasiado rápida de la presión intraocular50. El tratamiento en el glaucoma de aparición tardía es con medicamentos tópicos. Los supresores acuosos y los que aumentan el flujo de drenaje uveoescleral tienden a ser los más efectivos59,60. Los recién nacidos con riesgo de glaucoma (MVO frontal, periocular) deben de ser evaluados por oftalmología cada 3 meses durante los primeros años de vida, incluso si no se encuentra ninguna evidencia de glaucoma, se recomiendan exámenes anuales de por vida14.
Por último es importante controlar el crecimiento y los síntomas de disfunción tiroidea en los pacientes con SSW para detectar déficits de hormona del crecimiento e hipotiroidismo2,35.
PronósticoEl pronóstico en pacientes con SSW, depende principalmente de la edad de instauración de los síntomas neurológicos. El inicio a una edad precoz de las convulsiones, la falta de respuesta al tratamiento anticonvulsivo, la extensión de la malformación leptomeníngea, su efecto en la perfusión de la corteza cerebral y la gravedad de la afectación ocular determinan el pronóstico.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



