


The skin is the largest and most exposed organ in the human body and the ideal place to look for signs that aid in the early diagnosis of systemic diseases with cutaneous effects. As the concepts that underpin our understanding of many of these diseases have evolved or expanded in recent years, there have also been changes in the criteria we use for early diagnosis, including our approaches to skin biopsy and dermatopathologic evaluation. This review focuses on some of the systemic processes with skin manifestations for which our basic understanding has changed most in recent decades.
La piel es el órgano más extenso y más expuesto del cuerpo humano. Ello implica un magnífico terreno para el diagnóstico precoz de las enfermedades sistémicas que cursan con afectación sistémica y para las cuales, la piel se vuelve un marcador diagnóstico. Las bases conceptuales así como los criterios diagnósticos de muchas de estas entidades se han visto modificados o ampliados en los últimos años, con lo que la aproximación a la biopsia cutánea y la evaluación de los signos dermatopatológicos útiles en el diagnóstico precoz, han variado también. En esta revisión intentamos hacer un enfoque de algunos de los procesos sistémicos con repercusión cutánea que más han variado conceptualmente en las últimas décadas.
The skin is the largest and most exposed organ of the body. As a result, it is also the first to manifest many systemic conditions.
A review of all the systemic diseases that have skin manifestations would be impossible even in a book chapter. In the present article, we will focus on some recent or novel aspects of certain multiorgan diseases with skin involvement, as well as management of the best characterized of these in recent years, with a new conceptual framework for many of them.
Pathological Focus of Autoinflammatory DiseasesAutoinflammatory diseases are chronic, heterogeneous disorders arising from an abnormal phenotype in innate immunity.1 This results in a defect in the inflammasomes (multiprotein oligomers of myeloid cells), in turn causing an excessive inflammatory response, regulated by interleukin 1, to a diverse group of triggers.
Theoretically, these disorders are not associated with infectious, allergic, autoimmune, or immunodeficient processes. However, according to current thinking, most autoimmune and autoinflammatory disorders are actually mixed entities on a continuous spectrum.
Originally, 3 main types of autoinflammatory diseases were identified: Sweet syndrome (aseptic febrile neutrophilic dermatosis), pyoderma gangrenosum, and hidradenitis suppurativa. Over the last few decades, several entities associated with vascular diseases, certain genetic mutations, vasculitis, myelodycrasia, hereditary disorders, and solid tumors have been added to the list. Diseases now considered autoinflammatory diseases include erythema elevatum diutinum, dermatosis/arthritis syndromes associated with intestinal diseases, erysipeloid conditions associated with familial Mediterranean fever, urticarial rash, and periodic syndromes associated with cryopyrin or tumor necrosis factor receptor.
Two entities that have been reclassified as autoinflammatory diseases are interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis, which are now considered to lie at either end of a continuous morphologic spectrum.2
To summarize, autoinflammatory diseases are distributed over a broad spectrum. At one end of the spectrum are rare pure monogenic autoinflammatory diseases (DITRA, DIRA, Blau syndrome, FMF, TRAPS, HIDS, PAPA…), while at the other are rare monogenic autoimmune diseases (ALPS, IPEX, APCED…). In between there is a whole range of diseases with a combination of autoimmune and autoinflammatory components but a predominance of one or the other. Predominantly autoimmune diseases include classic polygenic autoimmune diseases (rheumatoid arthritis, pernicious anemia, autoimmune thyroiditis, Addison disease, myasthenia gravis, ANCA vasculitis, systemic lupus erythematosus, Sjögren syndrome, type 1 diabetes mellitus, gluten intolerance). Autoinflammatory diseases include classic polygenic diseases such as Crohn disease, ulcerative colitis, gout, reactive arthritis unrelated to major histocompatibility complex, vasculitis without antibodies, and idiopathic uveitis. Diseases in the mixed group with both autoimmune and autoinflammatory components in equal measure include reactive arthritis, ankylosing spondyloarthritis, psoriatic arthritis, Behçet syndrome, and HLA-B27-associated uveitis.
Several new entities have been included in the group of monogenic autoinflammatory diseases, such as pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA), pyogenic arthritis, pyoderma gangrenosum, acne, and suppurative hidradenitis (PAPASH),3 pyoderma gangrenosum, acne, suppurative hidradenitis (PASH),4 and psoriatic arthritis, pyoderma gangrenosum, acne, and suppurative hidradenitis (PsPASH).
Skin manifestations in autoinflammatory diseases are frequent, and in many cases an essential part of diagnosis. These manifestations include urticarial and maculopapular rash, pustules, cold sores, pyoderma gangrenosum, granulomatous lesions, and erysipelas-like lesions. Although none of these are pathognomonic of a specific syndrome, neutrophilic infiltrate is common to all of them.
Innate immunity is governed by neutrophils, basophils, and eosinophils, and so it is not unexpected that polymorphonuclear neutrophils, with or without vasculitis, are one of the main pathologic markers of autoimmune diseases. Although the presence of such markers is often obvious in a routine preparation with hematoxylin-eosin, special techniques, such as myeloperoxidase, may at times be required for diagnosis.5 This is because the neutrophil component is fragile and neutrophils may appear flattened, fragmented, or masked by more resistant cells such as lymphocytes, and so may resemble chronic infiltrate instead of an acute one.
Some studies suggested that neutrophilic infiltrates in autoinflammatory diseases are clonal or pseudoclonal,6 although this line of investigation has been subject of controversy in subsequent years.
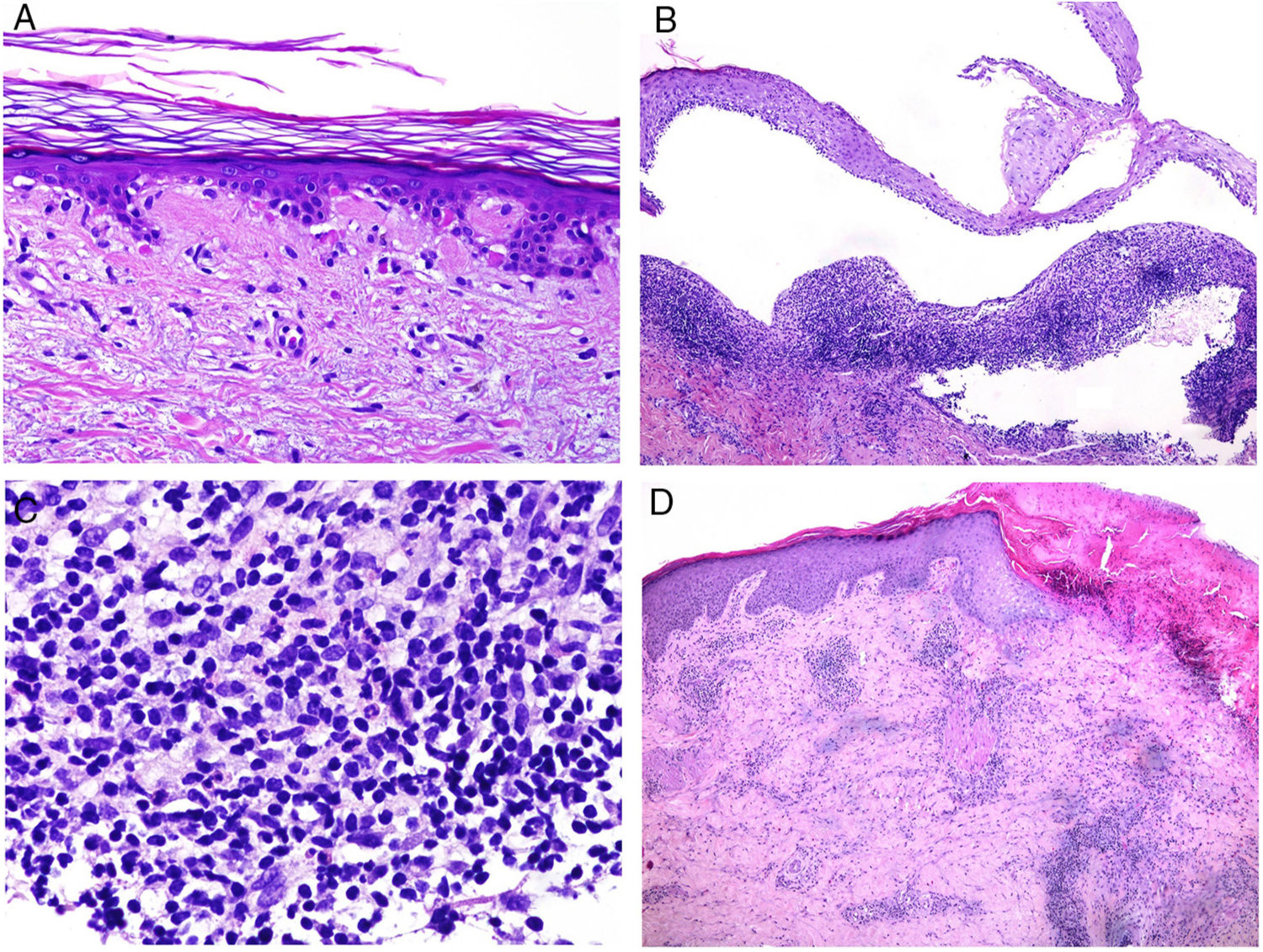
Neutrophilic infiltrates can be found in different layers of the skin, depending on the entity. Thus, they are preferentially epidermal in pustular psoriasis, generalized exanthematous pustulosis (Fig. 1), keratoderma blennorrhagicum, subcorneal pustular dermatosis (Sneddon-Wilkinson), IgA pemphigus, amicrobial pustulosis of the folds, infantile acropustulosis, and transient neonatal pustulosis. By contrast, the neutrophilic infiltrate is essentially dermal in entities such as Sweet syndrome, pyoderma gangrenosum, Behçet disease, arthritis-dermatosis associated with inflammatory bowel disease, neutrophilic eccrine hidradenitis, rheumatoid neutrophilic dermatitis, neutrophilic urticaria, Still disease, erythema marginatum, and hereditary periodic fever syndrome. Even within this last group, both the density and the depth of the dermal infiltrate may vary, being more superficial in Sweet syndrome—see for example Fig. 1B,C—while presentation may be deeper in Behçet disease or infiltrate the entire dermis and penetrate the hypodermis in pyoderma gangrenosum.
![A, Preferentially epidermal neutrophilic infiltrate in generalized exanthematous pustulosis (hematoxylin and eosin [H&E] ×100). B, Diffuse dermal infiltrate rich in mature polymorphonuclear neutrophils with intense subdermal edema and areas of bleeding in the papillary dermis (H&E ×20). C, At this magnification, polymorphonuclear neutrophils, as well as karyorrhexis, can be seen in greater detail (H&E ×200).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "A, Preferentially epidermal neutrophilic infiltrate in generalized exanthematous pustulosis (hematoxylin and eosin [H&E] ×100). B, Diffuse dermal infiltrate rich in mature polymorphonuclear neutrophils with intense subdermal edema and areas of bleeding in the papillary dermis (H&E ×20). C, At this magnification, polymorphonuclear neutrophils, as well as karyorrhexis, can be seen in greater detail (H&E ×200).")
A, Preferentially epidermal neutrophilic infiltrate in generalized exanthematous pustulosis (hematoxylin and eosin [H&E] ×100). B, Diffuse dermal infiltrate rich in mature polymorphonuclear neutrophils with intense subdermal edema and areas of bleeding in the papillary dermis (H&E ×20). C, At this magnification, polymorphonuclear neutrophils, as well as karyorrhexis, can be seen in greater detail (H&E ×200).
Still disease is a systemic inflammatory disorder of unknown etiology that presents clinically with peaks of intermittent fever, arthralgia, generalized swollen lymph nodes, sore throat, splenomegaly, and altered liver function.7 At times, the condition can be accompanied by serositis, myopericarditis, interstitial lung disease, and neurological involvement.7
The main skin manifestation of Still syndrome is evanescent rash comprising a salmon-pink maculopapular rash.8 However, atypical forms have been reported, with or without characteristic rash.9 Systemic symptoms may be concomitant with rash, precede rash by weeks or even years, or present after rash.
Within the atypical manifestations, the most frequent are persistent papules or plaques,10–12 (at times with linear or reticular distribution),13 urticarial reactions,14,15 prurigo pigmentosa-like reactions,16 generalized persistent nonpruritic erythema, or vesiculopustular eruption.9
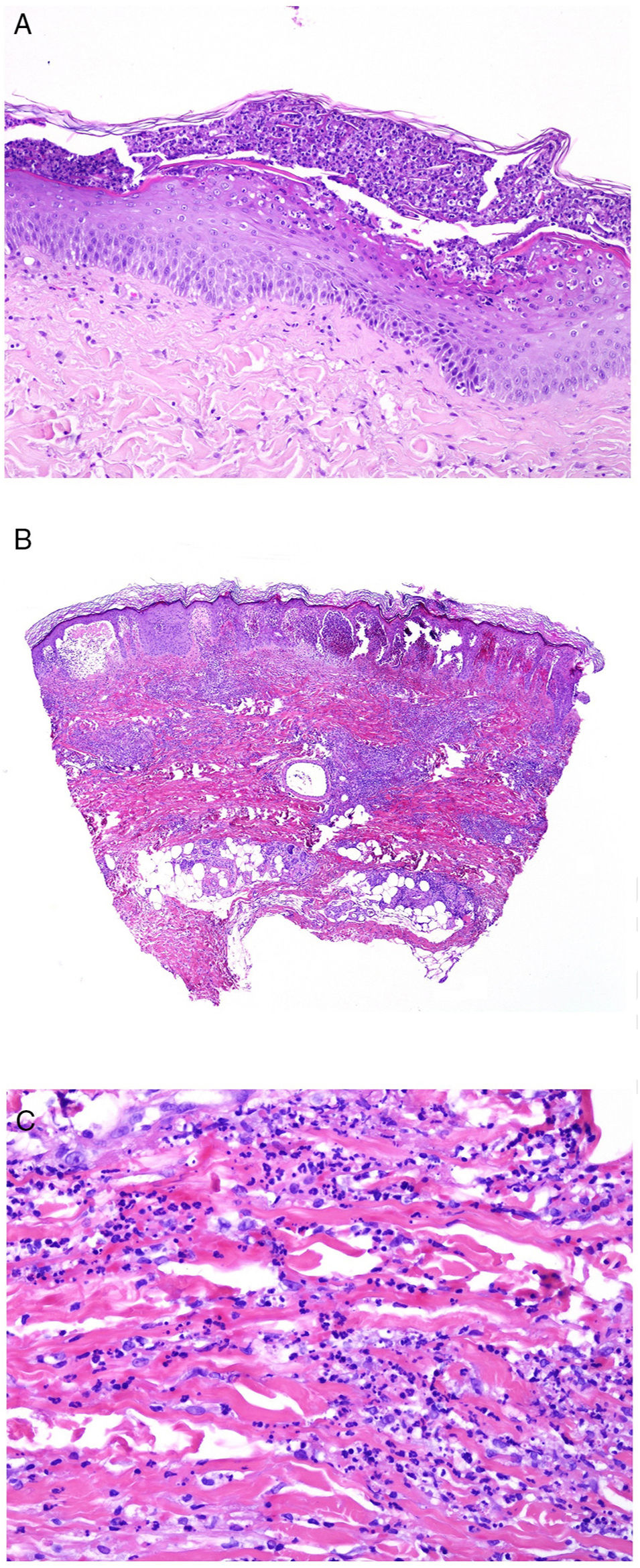
The pathologic presentation of classic evanescent rash in Still disease is nonspecific, with a superficial infiltrate that includes neutrophils, above which the epidermis appears preserved (Fig. 2A,B).
![A, Biopsy of an evanescent rash in a patient with Sill disease, showing a superficial perivascular lymphohistiocytic inflammatory infiltrate (hematoxylin and eosin [H&E] ×100) with some neutrophils (B, H&E ×400). C, In biopsies of patients with chronic Still disease, a superficial perivascular inflammatory infiltrate is still evident with some neutrophils (H&E ×100), but necrotic keratinocytes are also observed in the upper parts of the epidermis (D, H&E ×400).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "A, Biopsy of an evanescent rash in a patient with Sill disease, showing a superficial perivascular lymphohistiocytic inflammatory infiltrate (hematoxylin and eosin [H&E] ×100) with some neutrophils (B, H&E ×400). C, In biopsies of patients with chronic Still disease, a superficial perivascular inflammatory infiltrate is still evident with some neutrophils (H&E ×100), but necrotic keratinocytes are also observed in the upper parts of the epidermis (D, H&E ×400).")
A, Biopsy of an evanescent rash in a patient with Sill disease, showing a superficial perivascular lymphohistiocytic inflammatory infiltrate (hematoxylin and eosin [H&E] ×100) with some neutrophils (B, H&E ×400). C, In biopsies of patients with chronic Still disease, a superficial perivascular inflammatory infiltrate is still evident with some neutrophils (H&E ×100), but necrotic keratinocytes are also observed in the upper parts of the epidermis (D, H&E ×400).
In contrast, in persistent lesions, necrotic keratinocytes are observed, either in isolation or grouped in the upper parts of the epidermis, along with a superficial perivascular lymphohistiocytic infiltrate (at times also interstitial), which may be accompanied by neutrophils and eosinophils (Fig. 2C,D).10 This pathologic pattern is also the one observed when persistent plaques present a reticulated clinical pattern.13
Necrotic keratinocytes have been described in the literature as a source of diagnostic error, confused with erythema multiforme.17 However, erythema multiforme is an interface dermatitis in which at least some vacuolization of the basal layer is expected and in which necrotic keratinocytes are preferentially seen in the basal layer, although some may be present in higher layers. On the other hand, these are located in the upper layers of the epidermis in Still syndrome. Nevertheless, we should remember that on rare occasions, Still disease can show a pathologic pattern of vacuolar degeneration of the basal layer with subcorneal or intracorneal pustules.18
In some cases with clinical presentation of persistent brownish-red papules and plaques, biopsies have shown mucin deposits in the reticular dermis,19 a finding that has also been reported in cases of linear maculopapular rash.20
Urticariform clinical manifestations show typical pathologic characteristics of urticaria, with dermal edema and perivascular infiltrate (which is sometimes also interstitial), rich in neutrophils, without vasculitis.14
Another histopathologic pattern seen in atypical cases (mainly those that present with persistent diffuse pruritic erythema) is edema of the mid- and upper dermis with perivascular infiltrate of mononuclear predominance in which some neutrophils are observed.
The clinical description in the literature of prurigo pigmentosa-like lesions corresponds in biopsy to areas of parakeratosis and eosinophilic spongiosis in an acanthotic epidermis, isolated dyskeratotic cells in the epidermis, and a superficial perivascular dermal infiltrate comprised of neutrophils, eosinophils, and lymphocytes.16
Some atypical cases can present spongiosis with dermal edema accompanied by perivascular dermal infiltrate. These cases usually present clinically as vesiculopustular rash.
Finally, in the literature, some cases have been reported with oral involvement, with brownish macules on the internal oral mucosa of the lips and on the tongue.21 In these cases, biopsy has shown chorionic edema with a perivascular infiltrate rich in neutrophils.
Concept of IgG4-Related DiseaseIgG4-related disease is an entity described only relatively recently that presents with skin and systemic manifestations.22 In fact, it can affect almost any organ, although the most frequently involved are the pancreas and salivary and lacrimal glands.
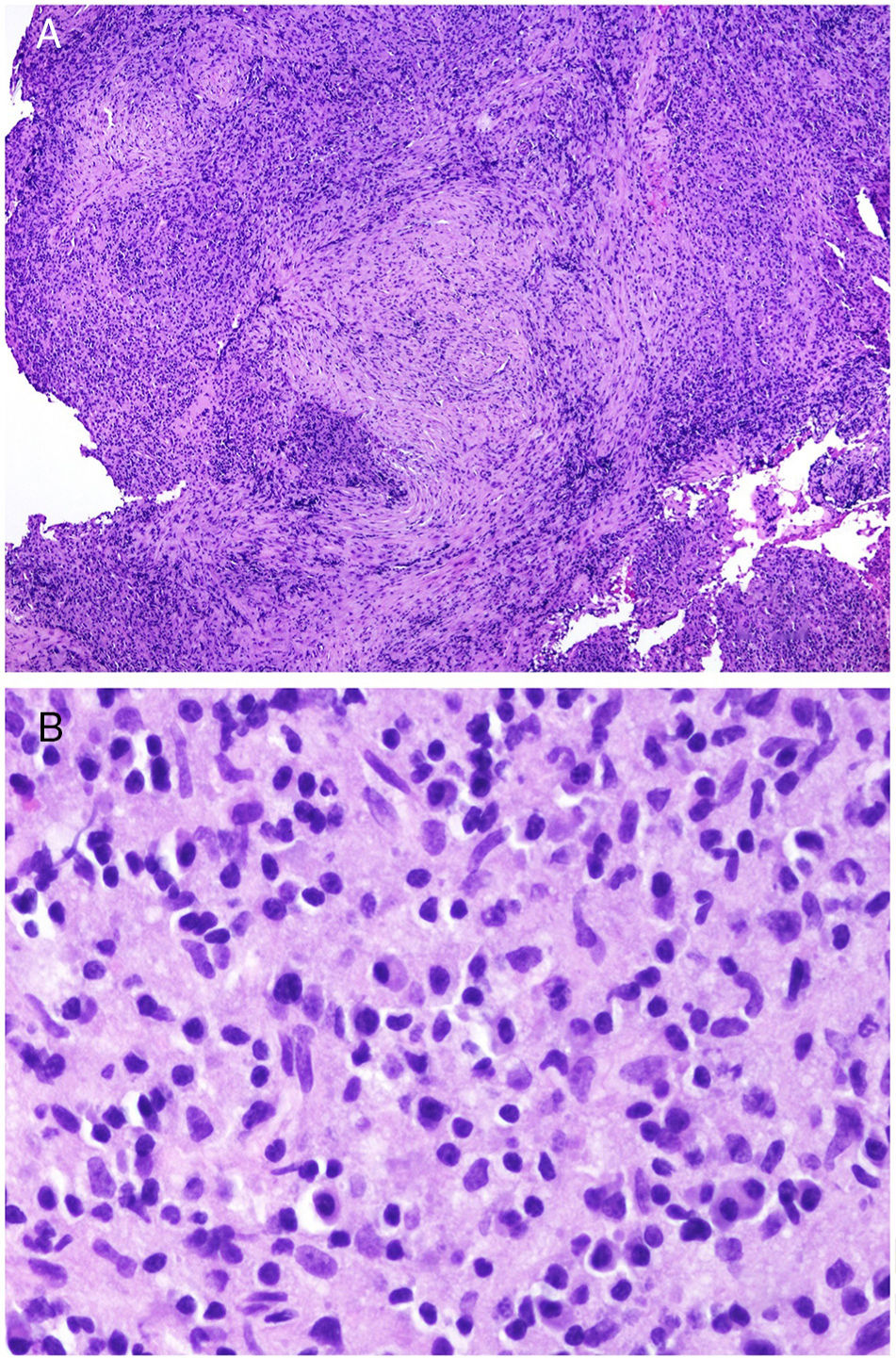
The pathologic marker for IgG-related disease is a lymphoplasmacytic infiltrate rich in IgG4+ cells along with fibrosis of storiform pattern (Fig. 3A,B).23 Specifically, the ratio of IgG4-IgG should be greater than 40% and IgG4+ plasma cell count should be higher than 10 per high-magnification field.24 Fibrosis, in contrast, does not usually appear until the disease is advanced and, therefore absence does not exclude diagnosis. The other frequent pathologic characteristic in this entity is obliterative phlebitis.
![A, At low magnification, Ig4-related disease should be suspected on finding an infiltrate rich in plasmatic cells coexisting with areas of concentric fibrosis (hematoxylin and eosin [H&E] ×40). B, Infiltrate rich in plasmatic cells in the case of IgG4-related disease (H&E ×400).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "A, At low magnification, Ig4-related disease should be suspected on finding an infiltrate rich in plasmatic cells coexisting with areas of concentric fibrosis (hematoxylin and eosin [H&E] ×40). B, Infiltrate rich in plasmatic cells in the case of IgG4-related disease (H&E ×400).")
However, this pathologic marker (richness of IgG4+ plasma cells) is controversial on its own outside a clinical context. This is because there are many entities that may present with IgG4+ cell enrichment. It has been suggested that skin diseases rich in plasma cells could actually be forms of IgG4+ related disease, as is the case for Rosai-Dorfman disease or angiolymphoid hyperplasia with eosinophilia.23 But not all the infiltrates rich in IgG4 are IgG4-related: abundant IgG4 plasma cells can be seen in chronic inflammation of the oral mucosa or in rheumatoid arthritis.25
There is evidence that IgG4 molecules are not inflammatory. Therefore, their presence seems more a secondary response to a primary process (which could be fibrosis) in individuals very likely with genetic predisposition.26–29 However, it could also be a phenomenon secondary to some immune inflammatory diseases or to chronic inflammatory processes that can present with infiltrates rich in IgG4+ plasma cells, without needing to meet the criteria for IgG4-related disease.30
From the clinical point of view, the disease presents with thickening and localized or diffuse masses or tumors in multiple organs.31 In the skin, the presentation is varied, in the form of macules, papules, plaques, nodules, purpura, blisters, or rash, with the most frequent site being the head and neck.32 Dermatologists should be alert to this diagnosis when these manifestations are associated with idiopathic pancreatitis, retroperitoneal fibrosis, aortitis, or previously diagnosed systemic IgG4-related disease.32
One of the most complicated differential diagnoses is systemic plasmacytosis, an entity most often occurring in Japan, which affects several organs including the skin, in the form of an eruption of reddish-brown papules preferentially distributed on the trunk.33 In biopsy, systemic plasmacytosis is characterized by a superficial and profound perivascular and periadnexal infiltrate of mature plasma cells with polytypical expression of immunoglobulins. Given that at times, the IgG4 subpopulation is prominent in this infiltrate, it has been suggested that at least in some cases, these could be forms of IgG4-related disease.34–36
Finally, we point out that IgG4-related disease is a recently described entity and so its causes and pathogenesis are not completely understood. This is probably a heterogeneous group of conditions that will likely soon be characterized.37
New Advances in Biopsy of LupusFrom the clinical point of view, lupus erythematosus with skin involvement can be divided in to 2 main groups: one group with mainly skin involvement (which basically includes discoid lupus erythematosus and the subacute form) and systemic lupus (for example, the acute form), which also affects internal organs.
A lengthy debate has considered whether there are histopathologic signs suggestive of systemic lupus erythematosus, and also whether there are forms of lupus confined exclusively to the skin, without any systemic impact. Progression to systemic forms in patients with chronic discoid lupus erythematosus indicates that there is no clear separation between the 2 groups. The distinction is, however, maintained because of the low risk of systemic progression of many of the cutaneous forms of lupus.
One of the forms of lupus that was considered as a primary cutaneous form without any systemic impact is lupus erythematosus tumidus, which is considered a separate entity.38 However, recently, a case of lupus erythematosus tumidus was reported in which conversion occurred from discoid lupus and which supports the interchangeability of the different variants of lupus.39 In contrast, other authors suggest that lupus should be considered a systemic diseases with variable impact on different organs, including the skin.40
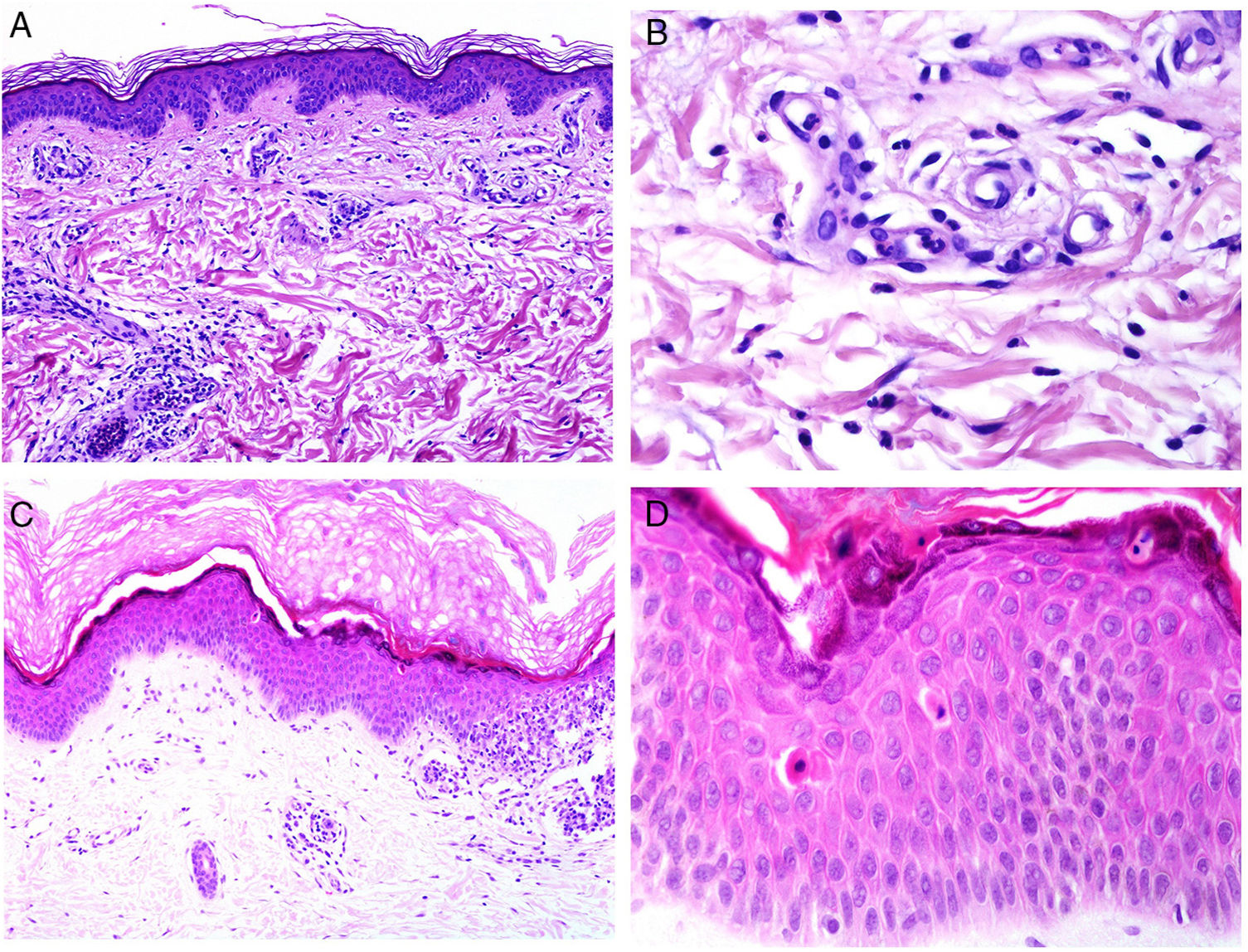
From the pathologic point of view, there are changes that are interpreted as signs of chronic or acute disease. Thus, while systemic lupus usually shows little or no epidermal atrophy (Fig. 4A), very little vacuolar degeneration of the basal layer (Fig. 4B), absence of thickening of the basal layer, minimal horny plugs, minimal dermal edema, and minimal periadnexal infiltrate (Fig. 4C); all these features are prominent in discoid lupus erythematosus (Fig. 4D).41
![Skin involvement in a case of lupus with systemic manifestations. Absence of epidermal atrophy (A, hematoxylin and eosin [H&E] ×100). There is very little vacuolar degeneration of the basal layer and no evidence of thickening of the basal membrane (B, H&E ×400). The dermal inflammatory infiltrate is discrete and may contain neutrophils (C, H&E ×200). D, In contrast, discoid lupus erythematosus shows infundibular dilatations, prominent horny plugs, and marked periadnexal infiltrate (H&E ×20).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "Skin involvement in a case of lupus with systemic manifestations. Absence of epidermal atrophy (A, hematoxylin and eosin [H&E] ×100). There is very little vacuolar degeneration of the basal layer and no evidence of thickening of the basal membrane (B, H&E ×400). The dermal inflammatory infiltrate is discrete and may contain neutrophils (C, H&E ×200). D, In contrast, discoid lupus erythematosus shows infundibular dilatations, prominent horny plugs, and marked periadnexal infiltrate (H&E ×20).")
Skin involvement in a case of lupus with systemic manifestations. Absence of epidermal atrophy (A, hematoxylin and eosin [H&E] ×100). There is very little vacuolar degeneration of the basal layer and no evidence of thickening of the basal membrane (B, H&E ×400). The dermal inflammatory infiltrate is discrete and may contain neutrophils (C, H&E ×200). D, In contrast, discoid lupus erythematosus shows infundibular dilatations, prominent horny plugs, and marked periadnexal infiltrate (H&E ×20).
Moreover, improved understanding in recent years has led to very different pathogenic scenarios of cutaneous lesions for systemic and localized forms. These 2 scenarios are, however, dynamic and may change over the course of a patient's lifetime according to external and internal factors.
Thus, migration of cytotoxic T cells and natural killer cells from circulation to the skin is characteristic of cutaneous forms but is not seen in systemic ones.41 Likewise, plasmacytoid dendritic cells, which have turned out to be very useful as a diagnostic marker of lupus, are usually abundant in cutaneous forms of lupus but not present to such an extent in skin biopsies of patients with systemic lupus, in which cells are preferentially confined to circulation. The opposite occurs with regulatory T cells, which appear very much reduced in skin biopsies of systemic lupus, but are not so reduced in lupus with skin involvement only.41
Plasmacytoid dendritic cells produce an environment with an excess of interferon alfa and beta, and these are not easy to measure in skin in everyday practice. However, myxovirus-resistant protein (MxA) seems to be a good alternative marker of levels of interferon in skin and peripheral blood.42
Some of the pathologic signs of biopsies of lupus erythematosus may be of prognostic use, in addition to supporting diagnosis. For example, there is a close relationship between the number of apoptotic keratinocytes and lupus erythematosus activity (Fig. 5A).41 There is greater apoptotic activity in subacute lupus and in the systemic form, 2 of the forms in which greatest systemic activity is observed.43 Furthermore, accumulation of apoptosis seems to be crucial as a trigger for skin lesions,41 contributing to inflammation and immune processes on release of proinflammatory molecules such as interferon alfa, interleukin 1, and HMGB1.44
![A, Numerous apoptotic figures in the basal epidermal layer (hematoxylin and eosin [H&E] ×200). The number of apoptotic keratinocytes has been correlated with lupus activity. B, Subepidermal detachment in a case of bullous lupus (H&E ×40). C, Presence of numerous polymorphonuclear cells in chronic infiltrate from a patient with bullous lupus (H&E ×400). D, Perivascular infiltrate in an ulcerated lesion corresponding to skin involvement in Kikuchi disease (H&E ×40).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr5.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "A, Numerous apoptotic figures in the basal epidermal layer (hematoxylin and eosin [H&E] ×200). The number of apoptotic keratinocytes has been correlated with lupus activity. B, Subepidermal detachment in a case of bullous lupus (H&E ×40). C, Presence of numerous polymorphonuclear cells in chronic infiltrate from a patient with bullous lupus (H&E ×400). D, Perivascular infiltrate in an ulcerated lesion corresponding to skin involvement in Kikuchi disease (H&E ×40).")
A, Numerous apoptotic figures in the basal epidermal layer (hematoxylin and eosin [H&E] ×200). The number of apoptotic keratinocytes has been correlated with lupus activity. B, Subepidermal detachment in a case of bullous lupus (H&E ×40). C, Presence of numerous polymorphonuclear cells in chronic infiltrate from a patient with bullous lupus (H&E ×400). D, Perivascular infiltrate in an ulcerated lesion corresponding to skin involvement in Kikuchi disease (H&E ×40).
One of the uncommon variants of lupus is the blistering form, which has recently been studied in a larger series of patients.45 The study showed that this should be considered a systemic form, associated with lupus nephritis in half of the cases. From the pathologic point of view, in addition to subepidermal detachment (Fig. 5B), of particular interest is the infiltrate rich in polymorphonuclear neutrophils (Fig. 5C).45 The good response obtained with dapsone in 90% of patients is therefore not surprising.45
There are also entities in which the classification as a form of lupus erythematosus is disputed. One of these is antiphospholipid syndrome, often overdiagnosed as lupus erythematosus and, as a result, inappropriately treated. To avoid this, some authors have suggested including the requirement for at least 1 criterion of systemic lupus erythematosus before making a diagnosis of lupus in patients with antiphospholipid syndrome.46
As in other diseases with a predominantly autoimmune component, lupus erythematosus has occasionally been associated with entities supposedly of autoinflammatory origin, such as hidradenitis supperativa.47
Recently, the possibility has arose of using ex vivo confocal fluorescent microscopy during the operation for a rapid diagnosis of inflammatory diseases, including lupus, with the aim of reducing delays in treatment.48
Skin manifestations of lupus are accompanied by increased skin carcinogenesis for a variety of reasons, including the presence of chronic scarring, persistent inflammation, sensitivity to ultraviolet radiation, and use of immunosuppressants for therapeutic management.49 However, both clinical observations of colocalization of multiple independent skin cancers on areas of active inflammation in patients with discoid lupus erythematosus in follow-up for several years, as well as murine experimental models, led to the conclusion that carcinogenesis is closely related to inflammation and that suppression or limitation of this leads to a considerable reduction in the risk of developing tumors.49
Lupus and Kikuchi-Fujimoto DiseaseKikuchi-Fujimoto disease (histiocytic necrotizing lymphadenitis) is an idiopathic disorder. The most widely accepted etiopathogenic hypothesis is the viral/autoimmune one.50 This is based on clinical findings (self-limiting course), laboratory findings (atypical lymphocytes in peripheral blood and increased cytokines, interferon alfa, and interleukin 6),51 and pathologic ones (predominance of T-CD8 lymphocytes in the infiltrate with apoptosis and necrotic background).52–54 This can be considered either an autoimmune disease or an exaggerated immune response to viral infection. Although the process usually resolves by itself, relapses may occur, sometimes many years after the first episode.55
Kikuchi-Fujimoto disease has a disputed relationship with systemic lupus erythematosus and, sometimes, both entities are present in the same patient. In these cases, Kikuchi-Fujimoto disease may occur before, at the same time, or after the presentation of lupus.56 At other times, patients diagnosed with Kikuchi-Fujimoto disease meet a series of criteria for lupus in some of their episodes.57,58
In addition, the lymph node changes observed in patients with systemic lupus and patients with Kikuchi-Fujimoto disease overlap, and in many cases cannot be distinguished.58 One of the morphological findings that has been considered as providing greatest discrimination between lupus and Kikuchi-Fujimoto disease is the presence of hematoxylin bodies that mimic DNA aggregates and anti-DNA antibodies.59 These hematoxylin bodies have mainly been observed in lymph nodes but they may also be present in biopsies of other organs, such as bone marrow or skeletal muscle.60,61 However, they are not a frequent feature of skin biopsies from patients with lupus erythematosus.
In contrast, skin biopsy in cases of Kikuchi-Fujimoto disease show perivascular and interstitial lymphocytic infiltrate (Fig. 5D) with presence of histiocytoid CD163+ and CD68+ cells that express myeloperoxidase.62 The nature of this infiltrate is currently under investigation. Necrotic foci without mature neutrophils are characteristic, as well as dermal-epidermal junction lesions.59,63 These latter lesions are a sign common to lupus, as are also mucin deposits and panniculittis.59
Given this overlap, Kikuchi-Fujimoto disease may actually be an incipient form of lupus erythematosus64 with a self-limiting and self-resolving course over a period of weeks or months in most cases.55,65 There are then a minority of cases with progression to severe systemic forms of Kikuchi-Fujimoto disease, some of which may be fatal, with response only to early and aggressive immunosuppressive treatment as well as cases of overlap with or progression to systemic lupus erythematosus.
Skin Biopsy in Diseases of the Digestive TractProbably, the best known digestive disease with skin involvement is gluten intolerance, with herpetiform dermatitis as a skin manifestation. The presence of granular IgA in the papillary dermis is due to the formation of immune complexes between IgA with avidity for the skin and the transglutaminase enzyme.66
In 2006, Humbert et al.67 proposed classifying skin diseases associated with gluten intolerance into 4 large groups according to their pathogenic mechanism. Those with an allergic mechanism include atopic dermatitis68 and urticaria, whether acute or chronic.69 Psoriasis figures among the autoimmune diseases.70–73 Other associated entities include rosacea and aphthous stomatitis.74,75 Although the list of associations is quite a lot longer, many entities have been associated only sporadically or by chance.
Another large group of digestive conditions with skin involvement are inflammatory diseases of the large intestine, with cutaneous manifestations of the granulomatous or reactive type, but also at times associated with nutritional deficiencies or iatrogenic factors.76 Excluding the last 2 (nutritional or iatrogenic), skin involvement is observed in 14.9% of patients with inflammatory bowel disease (Crohn disease or ulcerative colitis).76
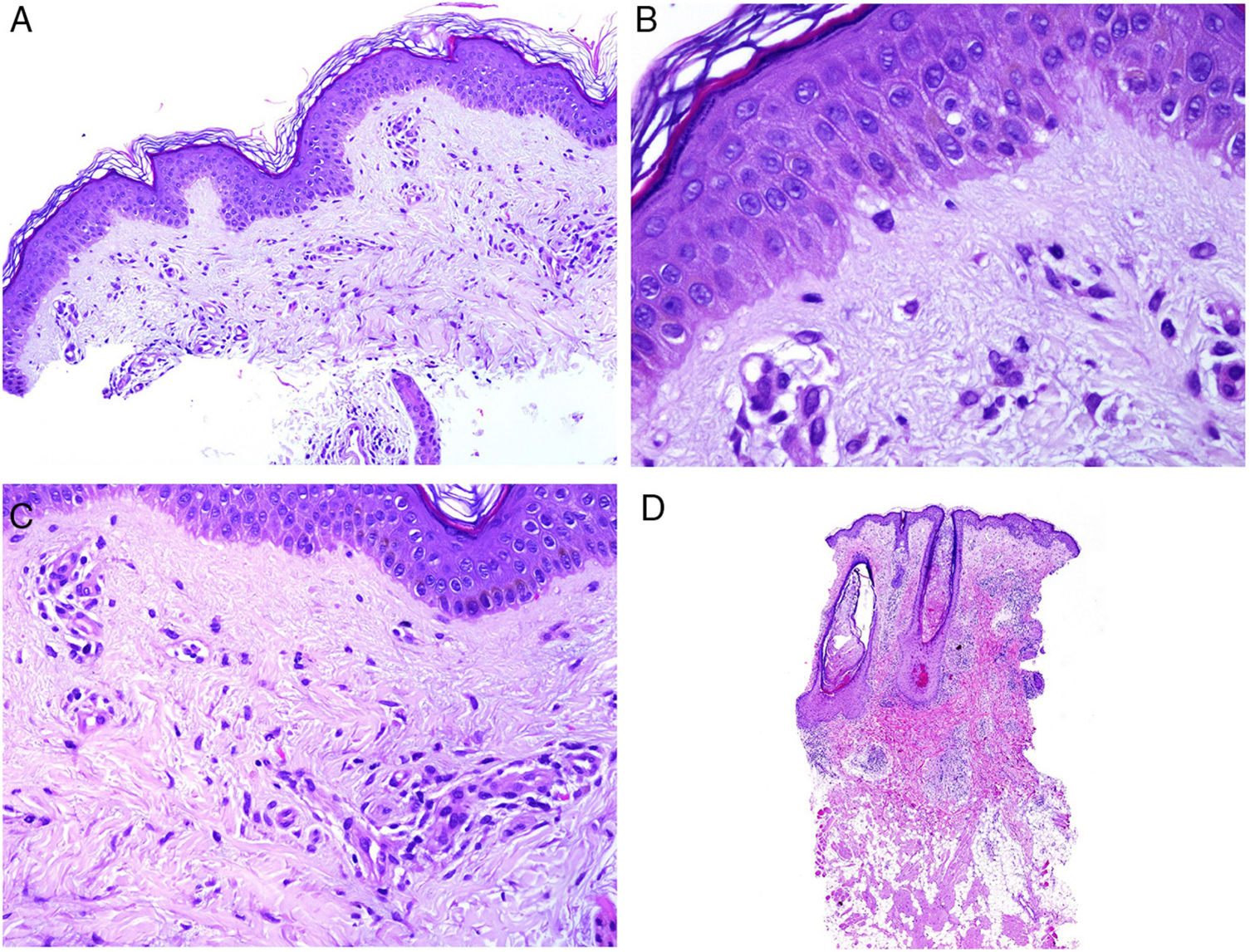
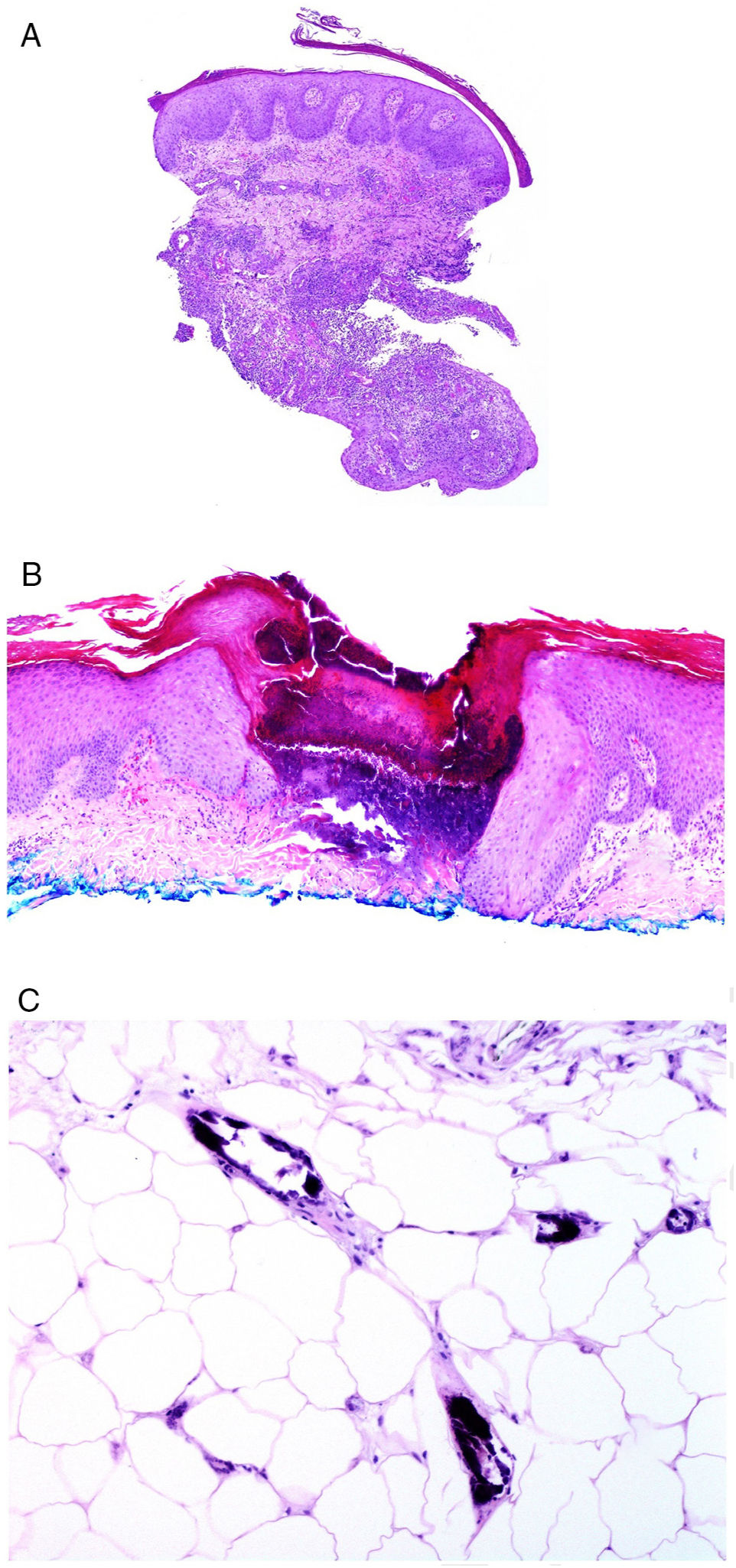
Involvement of skin closest to the gastrointestinal tract with noncaseating granulomas is considered specific to Crohn disease, sometimes accompanied by fistulas or abscesses (Fig. 6A).77 These same granulomatous manifestations can, however, be seen away from the anus and mouth, given rise to the term metastatic Crohn disease. Manifestations can appear even in patients whose gastrointestinal symptoms are well controlled with treatment.78
![A, Fistulous trajectory in biopsy of a metastatic Crohn lesion on the leg (hematoxylin and eosin [H&E] ×20). B, Perforating collagenosis with elimination of eosinophilic collagen fibers by a crater of necrotic material (H&E ×20). C, Calciphylaxis showing the calcified walls of small calliber subcutaneous vessels (H&E ×200).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr6.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "A, Fistulous trajectory in biopsy of a metastatic Crohn lesion on the leg (hematoxylin and eosin [H&E] ×20). B, Perforating collagenosis with elimination of eosinophilic collagen fibers by a crater of necrotic material (H&E ×20). C, Calciphylaxis showing the calcified walls of small calliber subcutaneous vessels (H&E ×200).")
A, Fistulous trajectory in biopsy of a metastatic Crohn lesion on the leg (hematoxylin and eosin [H&E] ×20). B, Perforating collagenosis with elimination of eosinophilic collagen fibers by a crater of necrotic material (H&E ×20). C, Calciphylaxis showing the calcified walls of small calliber subcutaneous vessels (H&E ×200).
In contrast, manifestations that do not show the granulomatous characteristic typical of Crohn disease but that probably correspond to skin involvement of the underlying autoinflammatory disorder are considered to be reactive. Many of these therefore show substantial dermal inflammatory infiltrate of polymorphonuclear neutrophils, such as pyoderma gangrenosum; Sweet syndrome; bowel-associated dermatosis-arthritis syndrome, pyodermatitis-pyostomatitis vegetans; synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome; and pyogenic arthritis, pyoderma gangrenosum, acne (PAPA) syndrome.77 Reactive manifestations also include erythema nodosum of characteristic histopathology with tendency for septal panniculitis.
The forms secondary to nutritional deficiencies are considered to be due mainly to lack of vitamin A, B12, and C.79 Finally, the drugs used in the treatment of inflammatory bowel disease are also responsible for a range of skin manifestations.
Hepatic autoimmune diseases such as primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis, show an association with different skin manifestations: the relationship with vitiligo has been demonstrated.80 Other probable associations include alopecia areata, psoriasis, pyoderma gangrenosum, and amyloidosis.81 In contrast to previous opinion, primary biliary cholangitis is not related to lichen planus but to amicrobial pustulosis of the skin folds.80
Recently, it has been reported that hepatitis virus E shows endothelial tropism able to guide T lymphocytes to clonality and trigger different types of lymphoma.82
Finally, we should remember that antiviral treatments in hepatitis are able to trigger systemic diseases such as lupus erythematosus83 and cryoglobulemia.84
Skin Biopsy in Renal DiseasesSkin manifestations associated with renal diseases can be classified in 3 categories: terminal renal disease, manifestations of uremia, and transplant-associated manifestations. Between 50% and 100% of patients with terminal renal disease have some type of skin manifestation.
Some of the skin manifestations are directly related to the primary underlying disease (Table 1), as is the case with diabetes, lupus erythematosus, Henoch-Schönlein purpura, granulomatosis with polyangiitis, polyarteritis nodosum, bacterial endocarditis, amyoloidosis,85 viral hepatitis, and systemic sclerosis, among others.
Systemic Diseases with Renal and Skin Involvement.
| Systemic DisorderSkin M | Manifestations |
|---|---|
| Diabetes mellitus | Skin infections |
| Necrobiosis lipoidica | |
| Diabetic dermopathy | |
| Bullosis diabeticorum | |
| Skin thickening syndrome | |
| Acanthosis nigricans | |
| Eruptive xanthomas | |
| Kyrle disease | |
| Manifestations of diabetic vasculopathy | |
| Manifestations of diabetic neuropathy | |
| Lupus erythematosus | Chronic form |
| Subacute form | |
| Acute form | |
| Bullous forms | |
| Tumidus form | |
| Mucosal ulcers | |
| Purpura | |
| Urticaria | |
| White atrophy | |
| Degos | |
| Livedo reticularis | |
| Thrombophlebitis | |
| Raynaud | |
| Cutis laxa | |
| Anetoderma | |
| Papular mucinosis | |
| Amicrobial pustulosis of the folds | |
| Perniosis | |
| Neutrophilic dermatosis | |
| Henoch-Schönlein | Purpura |
| Macular-papular or urticarial rash | |
| Blistering forms | |
| Granulomatosis with polyangiitis | Subcutaneous nodules |
| Ulcers | |
| Digital ischemia | |
| Vasculitis | |
| Panniculitis | |
| Polyarteritis nodosa | Livedo reticularis |
| Painful subcutaneous nodules | |
| Ulcers | |
| Purpura | |
| Skin necrosis | |
| Self amputations | |
| Subacute bacterial endocarditis | Janeway palmoplantar lesions |
| Osler subcutaneous digital nodules | |
| Roth retinal stains | |
| Conjunctival and mucosal petechia | |
| Nail splinter bleeding | |
| Amyloidosis | Periorbital purpura and in large folds |
| White papules | |
| Sclerodermiform lesions | |
| Nail dystrophy | |
| Alopecia | |
| Blistering lesions | |
| Macroglossia | |
| Viral hepatitis | Purpura |
| Ictericus | |
| Palmar erythema | |
| Lichen planus | |
| Urticarial vasculitis | |
| Cryoglobulinemia | |
| Erythema nodosum | |
| Erythema multiforme | |
| Behçet syndrome | |
| Porphyria cutanea tarda | |
| Hypertrichosis | |
| Sclerodermiform plaques | |
| Generalized granuloma annulare | |
| Porokeratosis | |
| Necrolytic acral erythema | |
| Polyarteritis nodosa | |
| Systemic sclerosis | Scleroderma |
| Chronic edema | |
| Sclerodactyly | |
| Scarring on the heel of the hand | |
| Raynaud | |
| Telangiectasias | |
| Cutaneous hypopigmentation and hyperpigmentation | |
| Alopecia | |
| Anhidrosis |
Among the effects associated with uremia are xerosis, pruritus, purpura, skin pigmentation, calciphylaxis, perforating diseases, porphyria and pseudoporphyria, sclerosis related to gadolinium accumulation, and alopecia. Different nail disorders, such as half-and-half nails (Linsday nails), Muehrcke lines, absence of lunula, nail paleness, and lamina coloration disorders can also be observed.86
Finally, the main skin manifestations related to renal transplant, in addition to rejection, are drug-associated disorders (Cushing-like changes, gingival hyperplasia, hair follicle changes), and those related directly to immunosuppression (infections of different types, skin tumors, porokeratosis).
One of the renal disorders with associated impact on the skin is Reed syndrome, with a tendency to renal cell carcinoma. Patients with the syndrome have associated cutaneous leiomyomas in which immune staining with fumarate hydratase is observed.87
Chronic renal failure is closely associated with perforating skin diseases, characterized by transepidermal elimination of different dermal components (elastic fibers, collagen, etc). Traditionally, these have been classified into 4 large groups: elastosis perforans serpinosa, reactive perforating collagenosis, perforating folliculitis, and Kyrle disease, with these 4 groups referred to by several authors with the overarching term acquired perforating dermatosis.
The pathogenesis of these perforating dermatoses is unknown although scratching is probably responsible in a significant number of patients, as shown by the fact that the lesions are usually much less numerous after treatment to mitigate pruritus.
From the pathologic point of view, perforating dermatosis presents as vertical invaginations of the epidermis in the form of a short channel with acanthotic epidermis at either end. The epidermis at the base of the canal may appear preserved or, at times, eroded. The channel contains basophilic degenerated material with neutrophils and connective tissue fibers (collagen in collagenosis, elastic fibers in elastosis, none in Kyrle disease, and even elastic and collagen fibers together) (Fig. 6B). In the case of elastosis perforans serpiginosa induced by D-penicillamine, the elastic fibers adopt a characteristic beady or lumpy-bumpy pattern.
Another crucial pathologic entity in the context of renal disease is calciphylaxis, a disease with a high mortality rate. Although the pathogenesis of this disease is unknown, calcification of capillary vascular walls and small and medium caliber vessels usually in subcutaneous sites (Fig. 6C) appears to be the main cause of chronic flow that leads to acute ischemic dermal and subcutaneous necrosis. This is accompanied by vascular thrombosis which probably translates into a state of hypercoagulability. As a result, dermal angioplasty is a frequent finding in pathologic study.
To demonstrate calciphylaxis, a deep punch biopsy is required to enable assessment of vessels in the hypodermis. However, sometimes, there is a more superficial histopathologic finding that is pathognomonic of calciphylaxis: stippled perieccrine calcifications. In addition, neural calcifications can also be seen.
Finally, we note that the presence of calciphylaxis does not necessarily imply renal disease: cutaneous histopathologic findings of calciphylaxis observed in patients with and without renal disease appear to be similar.
Skin Biopsy in Sarcoidosis: The Concept of Silica GranulomaOne of the most controversial aspects in the literature is the finding of sarcoidal granulomas in skin biopsy, and whether this bears any relation to systemic sarcoidosis.
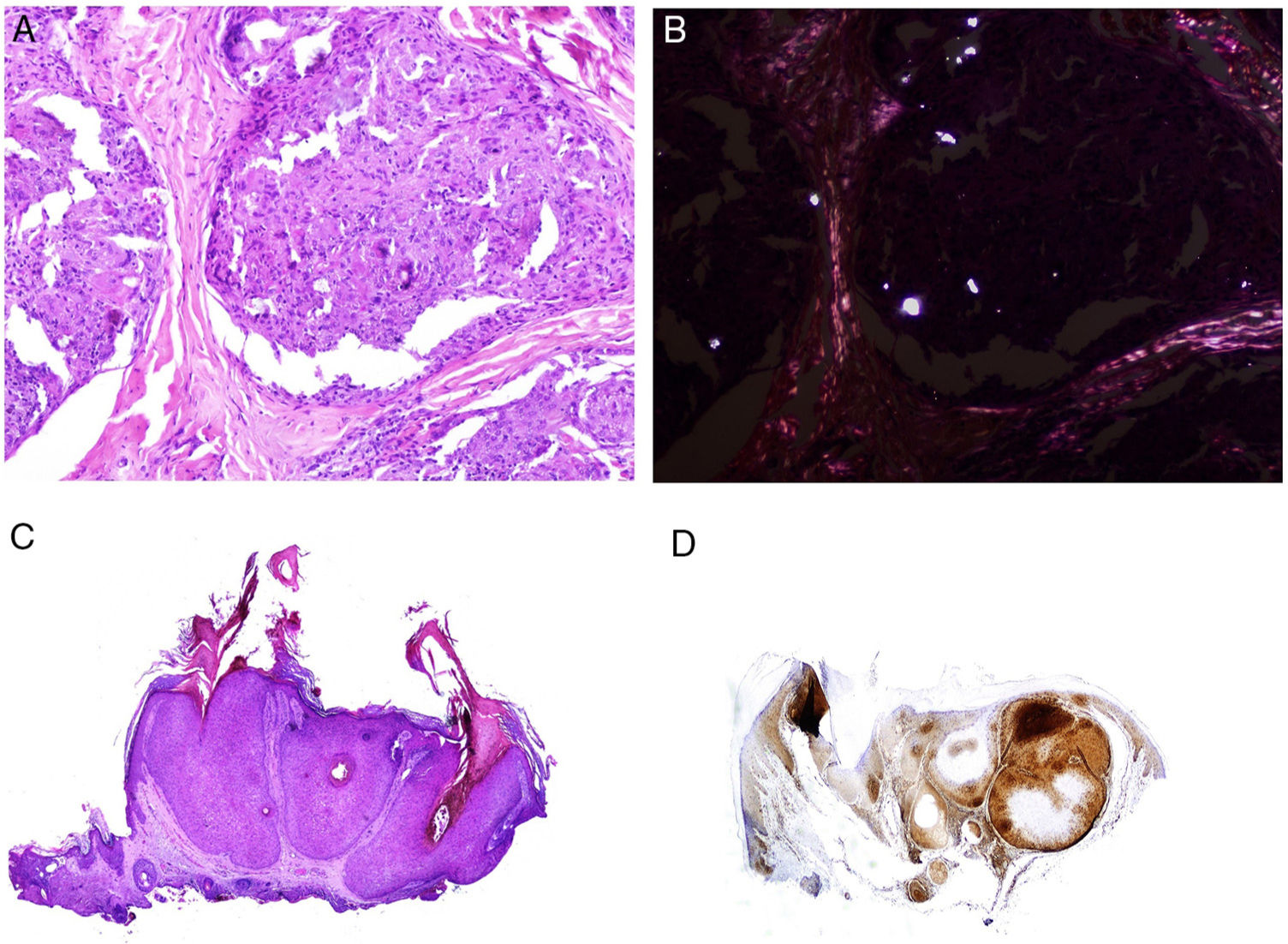
Many of these granulomas, when examined with polarized light, contain birefringent silica particles (Fig. 7A). This has given rise to the concept of silica granuloma.88 Very recently, these deposits of sarcoidal granulomas have been characterized physically and chemically, with crystalline silica present in central areas of granuloma and calcite in the periphery.89
![A, Sarcoid epithelioid granulomatous reaction in the dermis in a case of silica granuloma (hematoxylin and eosin [H&E] ×100). B, In the examination of sarcoid granulomas with polarized light, numerous intensely birefringent particles can be observed (H&E ×100, polarized light). C, Typical example of trichilemmoma (H&E ×20); even at this magnification, characteristic cytoplasmic cellular vacuolizations can be observed. D, Preservation of PTEN expression in a sporadic trichilemmoma, not associated with Cowden syndrome (PTEN × 30).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr7.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "A, Sarcoid epithelioid granulomatous reaction in the dermis in a case of silica granuloma (hematoxylin and eosin [H&E] ×100). B, In the examination of sarcoid granulomas with polarized light, numerous intensely birefringent particles can be observed (H&E ×100, polarized light). C, Typical example of trichilemmoma (H&E ×20); even at this magnification, characteristic cytoplasmic cellular vacuolizations can be observed. D, Preservation of PTEN expression in a sporadic trichilemmoma, not associated with Cowden syndrome (PTEN × 30).")
A, Sarcoid epithelioid granulomatous reaction in the dermis in a case of silica granuloma (hematoxylin and eosin [H&E] ×100). B, In the examination of sarcoid granulomas with polarized light, numerous intensely birefringent particles can be observed (H&E ×100, polarized light). C, Typical example of trichilemmoma (H&E ×20); even at this magnification, characteristic cytoplasmic cellular vacuolizations can be observed. D, Preservation of PTEN expression in a sporadic trichilemmoma, not associated with Cowden syndrome (PTEN × 30).
For some authors, the mere presence of silica rules out systemic sarcoidosis.90–92 In contrast, other studies have demonstrated that silica particles are frequent in cutaneous granulomas of patients with systemic sarcoidosis.93,94
Silica is an ubiquitous material in nature. It probably penetrates our skin at a very early stage in our lives, as a result of mild trauma during childhood, and has a long latency period.95 Indeed, it is perhaps surprising that granulomatous reactions to silica are not more common.
Some studies have demonstrated that particles of silica are as frequent in cutaneous sarcoidal granulomas in patients with sarcoidosis as in those without sarcoidosis.96 Furthermore, when skin biopsies are studied from patients with different diseases unrelated to sarcoidosis or granulomatous inflammation, silica particles are found in almost 40%.96
As a result, it has been suggested that although silica particles could be a trigger, a predisposition is necessary, for example in patients with sarcoidosis, and so these patients are more susceptible to developing granulomas in the face of certain stimuli.97 This mechanism has been corroborated by the description of similar granulomatous processes in patients with sarcoidosis as in processes associated with other types of particles, such as for example tattoos,98 mesotherapy,99 in-dwelling catheters,100 and, more recently, for example the development of sarcoidal granulomas in predisposed patients at venipuncture sites.101 A similar process has been reported in other organs in response to other types of particles (such as talc) in patients with a tendency to develop other types of granulomatosis, such as for example Crohn disease.102
Multiple Trichoepitheliomas and Cowden SyndromeCowden syndrome is a autosomal dominant genodermatosis with multiorgan involvement in which there is a risk of developing neoplasms, in particular breast, thyroid, and endometrial tumors.103 Clinical expression of the syndrome is variable, as are the causative mutations identified so far.104,105 The term is not derived from the clinicians who discovered the entity but from the patient in whom it was described, Rachel Cowden, who later died of breast cancer.106 Furthermore, the syndrome is characterized by multiple hamartomas at many sites, such as oral mucosa, skin, gastrointestinal tract, bone, eyes, central nervous system, and urinary tract.107
The cutaneous marker of Cowden syndrome is the presence of multiple facial trichilemmomas (more than 3) (Fig. 7C), which are considered pathognomonic of the syndrome. Other pathognomonic markers are acral keratosis, oral papillomatosis, and Lhermitte-Duclos disease.
Although the etiology is unknown, a mutation in the phosphatase and tensin homolog (PTEN) tumor suppressor gene is observed in up to 85% of cases. The function of the gene product, the PTEN protein, is not entirely characterized but involves modulation of the cell cycle and cell survival. Inactivation leads to growth and excessive cell survival.
It has been suggested that PTEN could be used as a immunohistochemical marker for Cowden syndrome, as this gene is not expressed in up to 83% of cases of the syndrome (Fig. 7D).108 However, absence of expression is not pathognomonic as it is lacking in between 3% and 13% of sporadic cases.108,109 In addition, there are forms of Cowden syndrome that are not associated with the PTEN mutation.110
Recently, a segmental presentation of storiform cholagenomas has been reported as part of the spectrum of the syndrome.111
Approach In Patients With Suspicion of Muir-Torre SyndromeLynch syndrome, also known as hereditary non-polyposis colorectal cancer, is an autosomal dominant condition associated with mutation of the germ line of DNA mismatch repair genes, responsible for between 5% and 10% of colorectal cancers. In 90% of colorectal cancers that develop in patients with Lynch syndrome, there is a relationship with the pathogenic mechanism of microsatellite instability (MSI). The microsatellites are repeated sequences of 1 to 6 base pairs in length that appear normally in our DNA. Of note is that although the length varies from one person to another, their length is constant in a given person. If there are defects in the DNA mismatch repair proteins, we find a variation in microsatellite length in an individual. This condition is not a direct cause of tumor risk but rather a consequence, a marker, and a warning of the incorrect functioning of the DNA mismatch repair system.
Patients with Lynch syndrome have a functional allele and a defective one for DNA mismatch repair. The syndrome develops after an incapacitating somatic mutation of the functional allele.112 As this pathway implies deficient repair of genetic errors in general, individuals with Lynch syndrome have a risk of developing other cancers at an early age, with endometrial cancer being the most frequent. To a lesser extent, there is also a risk of ovarian, stomach, small intestine, pancreatic, hepatobiliary tract, urinary tract, prostate, brain, and skin cancers.
The presentation of cutaneous tumors associated with MSI is known as Muir-Torre syndrome, which is therefore considered a variant of Lynch syndrome. Autosomal dominant transmission is observed in 59% of patients, with a high degree of penetrance and variable expression. The cutaneous tumors that develop most frequently are sebaceous tumors and keratoacanthomas. The former include adenoma sebaceum, sebaceoama, sebaceous carcinoma, and basal cell carcinoma with sebaceous differentiation. Nevus sebaceus of Jadassohn is not included among these tumors. Although sebaceous hyperplasia bears some relation to Muir-Torre syndrome, it is usually excluded from the list given that it frequently occurs in patients without the syndrome. Similar considerations apply to keratoacanthoma (both solitary and multiple presentation), spindle cell carcinoma, and multiple infundibular cysts, all associated with Muir-Torre syndrome but also frequently observed in the general population and so their usefulness as a marker is reduced. Nevertheless, some authors suggest investigating whether Muir-Torre syndrome is present in cases of multiple keratoacanthoma or keratoacanthoma with sebaceous differentiation. Recently, a subgroup of tumors has been described with a particular morphological pattern (carcinoid-like) which does not seem to be related to Muir-Torre sydnrome.113
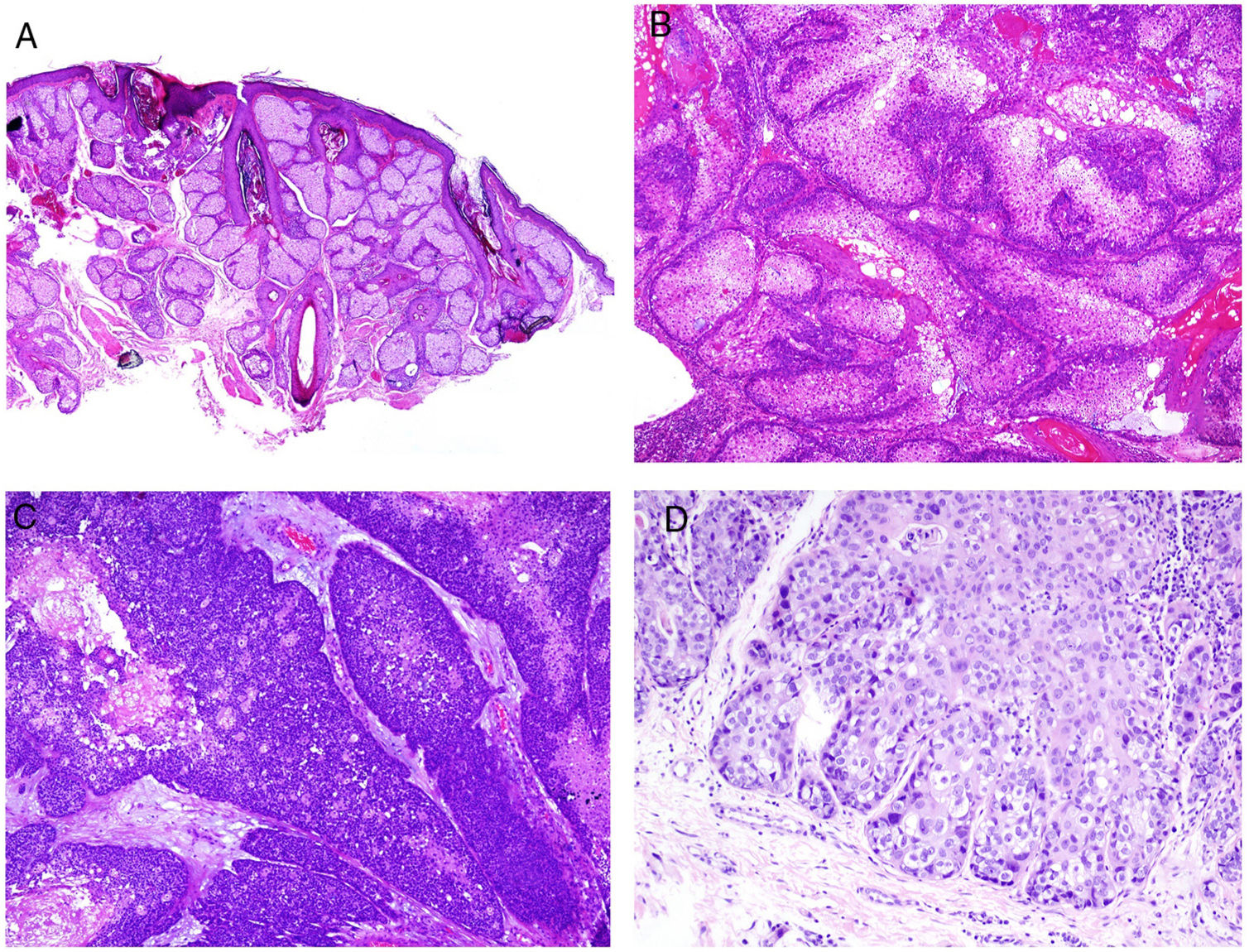
Adenoma sebaceum is the sebaceous neoplasm most frequently associated with Muir-Torre syndrome. This is a multilobed dermal tumor. Each lobe appears to be surrounded by a pseudocapsule of connective tissue and is made up primarily of multivacuolated cells with a mature sebaceous appearance (Fig. 8B). At the periphery of each lobe, several layers of basophilic germ cells can be observed (in contrast with sebaceous hyperplasia which, at most, shows 2 layers of germ cells; Fig. 8A). These latter cells may sometimes have a palisade arrangement, although this is not the most frequent. Unlike adenoma sebaceum, sebaceoma mainly consists of mature basaloid cells and does not usually have a lobed architecture (Fig. 8C). However, there is a morphological continuum between adenoma sebaceum and sebaceoma, and so some authors prefer to use the unifying term of sebomatricoma. It is important to note that both neoplasms generally have a symmetric architecture (unlike sebaceous carcinoma) and, from the cytological point of view, do not show nuclear pleomorphism (Fig. 8D). Necrosis is also an important indicator of malignancy in sebaceous neoplasms, unlike mitosis, which can be seen in abundance even in benign sebaceous tumors.
![A, Sebaceous hyperplasia (hematoxylin and eosin [H&E] ×20). Sebaceous Muire-Torre syndrome associated neoplasms. B, Adenoma sebaceum (H&E ×40). C, Sebaceoma (H&E ×40). D, Sebaceous carcinoma (H&E ×200).](https://static.elsevier.es/multimedia/15782190/0000011000000009/v2_201911260739/S1578219019302628/v2_201911260739/en/main.assets/gr8.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AlznLS2DX13Vc79dihNkcr0VDmf+2nqZboc/CWaVIgWo9Q+YxyKAlGPk87E2ZQoRY17Y0wxxGcEftFXakz81i+ll8lVNrjrGMcMBW8ZzY6NeIlJKSI4OS0jeJ1tRNfAfK+YAASYxer1VupIQSNNd2QDTpHzxCGEAx7SOHXEyoXcIrwZ3BaBkRujGkTXKk6TuoqooioJgz+P0jkC9DFXT+IU= "A, Sebaceous hyperplasia (hematoxylin and eosin [H&E] ×20). Sebaceous Muire-Torre syndrome associated neoplasms. B, Adenoma sebaceum (H&E ×40). C, Sebaceoma (H&E ×40). D, Sebaceous carcinoma (H&E ×200).")
Distinction should be made between the 3 aforementioned sebaceous neoplasms and basal cell carcinoma with sebaceous differentiation, which is a tumor with many of the classic basal cell attributes (peripheral palisading, stromal retraction, stroma with mucin, among others). Sebaceous differentiation is usually observed in the form of mature sebocytes in a variable quantity.
It should be clarified that sebaceous tumors associated with Muir-Torre syndrome often show ambiguous morphological characteristics that do not permit a clear classification as benign or malignant. However, this question is not usually critical if the neoplasm has been fully resected. Muir-Torre syndrome is an example of MSI.114,115
To sample this condition of risk, the microsatellites of the individual can be sequenced but this is costly and laborious for a screening process. There are other more practical alternatives. Specifically, determination of DNA mismatch repair protein expression. The National Cancer Institute recommends the inclusion of 5 markers for detection of individuals with MSI116: MutL homolog 1 (MLH1), MutS protein homolog 2 (MSH2), MutS homolog 6 (MSH6), Postmeiotic segregation increased 2 (PMS2), and MutL homolog 3 (MLH3). The genes that encode these proteins belong to the mismatch repair system. This is a very important system in evolutionary terms, and one that is highly preserved from bacteria to humans.
MLH1 and MSH2 defects are detected in 90% of cases of Lynch syndrome, while MSH6 defects are detected in 7%–10% and PMS2 defects in 5%.117 The association with MLH3 is very small (less than 3%) and so most laboratories do not include this marker in the study. Alternatively, Lynch syndrome can be the result of epigenetic alterations,112 or of sporadic somatic mutations in both alleles (rare).118
Interestingly, most patients with brain tumors show mutations in MSH2 with preservation of MLH1 and present a family history of brain tumors,119,120 and so this possibility should be investigated in clinical questioning when this phenotype is present.
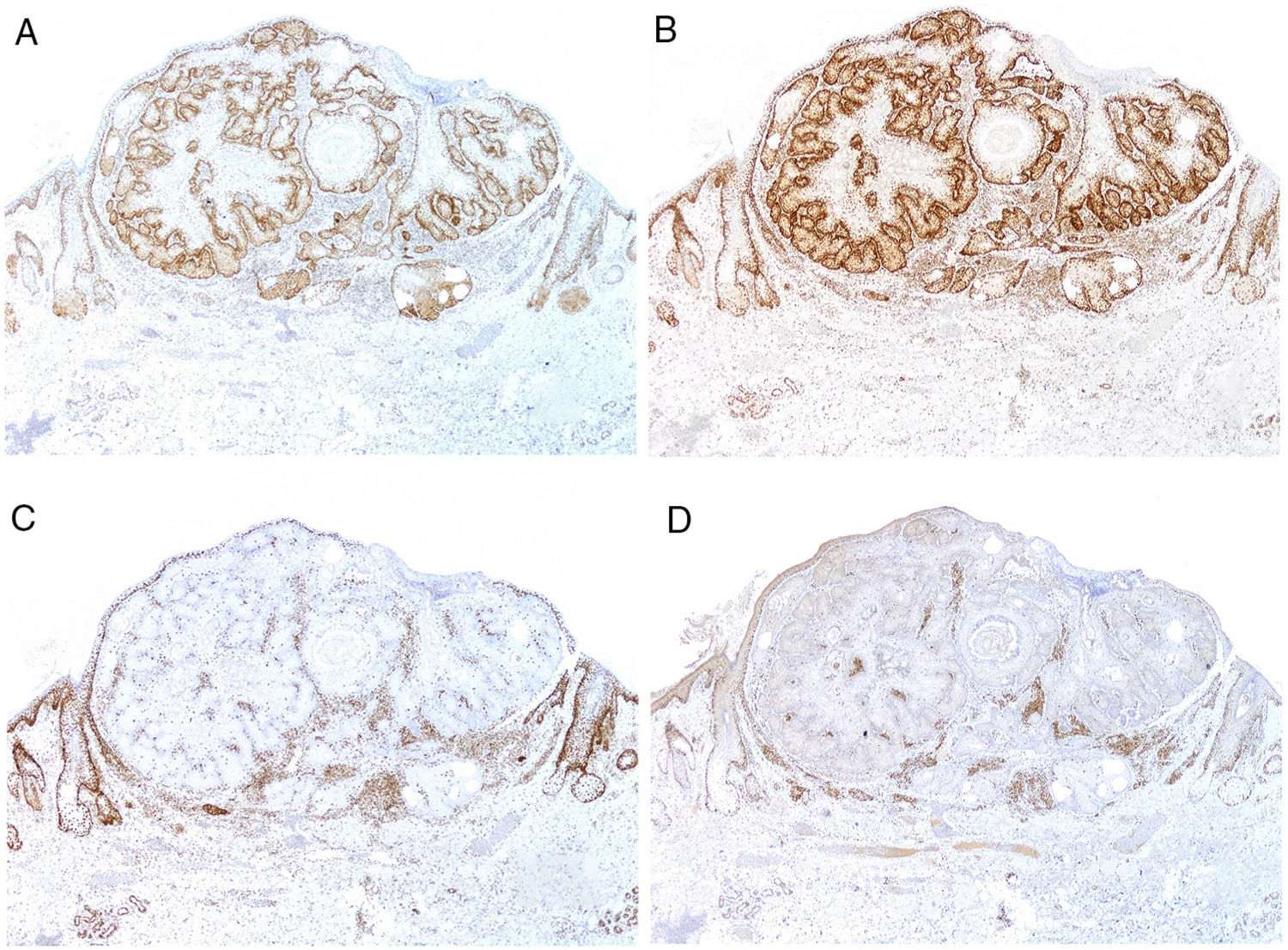
Currently, most hospitals perform a first systematic screening study in all colorectal, endometrial, and small intestinal carcinomas through immunohistochemical staining for MSH2, MSH6, MLH1, and PMS2 (Fig. 9), as well as for sebaceous adenomas, sebaceous carcinomas, sebaceomas, and basal cell carcinomas with sebaceous differentiation. It is still subject of debate whether keratoacanthomas or sebaceous hyperplasia should be included.
 and MLH1 expression (B, ×20) with loss of MSH2 expression (C, ×20) and MSH6 expression (D, ×20).")
Regardless of the result of this screening, patients with one of the aforementioned tumors should be questioned as to whether they meet the Bethesda or Amsterdam criteria or, alternatively, the Mayo Clinic risk score for Muire-Torre syndrome should be calculated.121 If this investigation is positive, a first step could be study of the mismatch repair markers by immunohistochemistry, and if these results are inconclusive, genetic and molecular analysis should be performed to rule out MSI.
The inactivity of some of the genes that encode mismatch repair proteins may also be due to hypermethylation of their promotor. In this case, the search for a mutation will be futile. Given that BRAF mutations are frequent in sporadic colorectal cancers but infrequent in those associated with Lynch syndrome, it has been suggested that study of these mutations would be a useful step prior to study of mismatch repair markers.122 However, study of the BRAF V600E mutation (the most frequently investigated by laboratories) showed wild-type BRAF was present in all sebaceous neoplasms associated with Muir-Torre syndrome investigated.123
The use of BRAF has also been suggested after immunohistochemistry for mismatch repair proteins in cases of loss of MLH1 expression not associated with MSH2, given the strong association between BRAF V600E mutation and methylation of the MLH1 promotor.124
The key question, and one subject to ongoing debate in the literature, is whether systematic study is required of all resected sebaceous tumors or only those associated with Muir-Torre syndrome. The most accepted approach is to only include study of adenoma sebaceum, sebaceoma, and sebaceous carcinoma. As mentioned above, the rationale for excluding sebaceous hyperplasia and keratoacanthomas from the study is largely based on the high frequency of presentation in the population without the syndrome. However, immunohistochemical study with the 4 mismatch repair markers is currently inexpensive, and so it would be justified to include at least sebaceous hyperplasia in these studies.
BAPoma ConceptBAP-1 is the protein associated with the BCRA-1 (BreastCancer-1) tumor suppressor gene. This gene encodes the mismatch repair protein BCRA-1. BAP-1 binds to BRCA-1 to form a repair complex with antitumor function.125
Defects in the BAP-1 gene can present as an autosomal dominant hereditary condition with as yet undetermined penetrance, in which affected individuals develop several types of somatic mutation of the functional allele,126 showing predisposition to uveal and cutaneous melanoma and also to mesothelioma, cholangiocarcinoma, renal carcinoma, and basal cell carcinomas.
BAP-1 negative nevi were described as a cutaneous marker for the syndrome. They present as multiple, papular lesions (between 5 and 50) usually during the second decade of life.126 The interesting point from the anatomopathologic point of view is that they can be identified by their morphological characteristics (although before the entity had been described, probably most if not all were classed as Spitz nevi). These are nevi that are preferentially dermal with extensive cytoplasm and prominent nucleoli.127 Immunohistochemical study for BAP-1 shows loss of nuclear positivity, leading to the paradox of using the term BAPomas when they actually do not express BAP. An alternative to this term is the eponymous Wiesner nevus, as Wiesner was the first author to describe the lesion.126,128
Likewise, an uncommon presentation is as a combined nevus in which one part that is negative for BAP-1 coexists with another part with another non-Spitzoid conventional BAP-1 positive part.129
Unlike in Spitz nevi, Wiesner nevi have BRAF mutations.130,131 However, as in almost all skin lesions, all information should be put into context, as not all melanocytic tumors with loss of BAP-1 are Wiesner nevi; conventional Spitz nevi have been reported with loss of BAP-1 expression.129
As with many other markers, Wiesner nevi do not always present in the context of familial disease but rather can present sporadically.
Recently, a multicenter study was published of the common clinical and dermoscopic characteristics of 48 Wiesner nevi from 31 patients.132 The authors concluded that the entity should be suspected in the presence of cupuliform papules with amorphous pinkish-brown areas and periphery that show either irregular globules-dots or an irregular lattice.
From the histopathological point of view, Wiesner nevi are symmetric under low magnification, although as the magnification is increased, these are clearly comprised of different types of cell population with predominance of epithelioid melanocytes with broad cytoplasm and atypical hyperchromatic nucleus. Melanocytic maturation is not observed, that is, the melanocytes of the deeper areas are not smaller or more monotone than those from the upper parts of the nevus. Frequently, the contrary paradox may even arise; there appears to be an inverse maturation process, with populations of very small cells in the upper parts, reflecting a certain chaotic structuring in these nevi. The melanocytic population can be accompanied by a moderately inflammatory infiltrate.
ConclusionsSkin manifestations in systemic inflammatory processes enable an early diagnosis of these entities, allowing subsequent appropriate therapy. A different approach in recent years with some of these manifestations has allowed better management, classification, and understanding of the underlying systemic inflammatory processes.
Conflicts of InterestThe author declares that he has no conflicts of interest.
Please cite this article as: Fernandez-Flores A. La biopsia cutánea en el contexto de la enfermedad sistémica. Eczema y urticaria en Portugal. 2019;110:710–727.



