





El tumor mixohialino inflamatorio de las áreas distales de las extremidades (TMHIADE) es un sarcoma de bajo grado de malignidad extremadamente infrecuente, con tendencia a la recurrencia local tras su extirpación quirúrgica, pero con un bajo potencial metastásico. Presentamos el caso de una mujer de 49 años que consultó por una tumoración asintomática de lento crecimiento en la zona pretibial derecha, que inicialmente sugirió el diagnóstico de lipoma. El estudio histopatológico mostró la presencia de un infiltrado inflamatorio polimórfico inmerso en una matriz mixoide e hialina. Entremezcladas entre las células inflamatorias existían varias poblaciones de células tumorales: en primer lugar, unas células atípicas de morfología fusiformes; en segundo lugar, unas células epitelioides bizarras, algunas de las cuales eran multinucleadas y recordaban a los virocitos o células de Reed-Sternberg; por último, unas células de citoplasma amplio multivacuolado que recordaban a los lipoblastos.
Estos hallazgos clínico-patológicos permitieron el diagnóstico de tumor mixohialino inflamatorio de las áreas distales de las extremidades. A pesar de que la tumoración se extirpó con márgenes quirúrgicos amplios, presentó una recidiva local tres meses después que fue tratada con nueva extirpación quirúrgica y radioterapia.
Inflammatory myxohyaline tumor of the distal extremities is an extremely rare low-grade sarcoma with a tendency to produce local recurrence after surgical excision, but with a low metastatic potential. We present the case of a 49-year-old woman with a slow-growing asymptomatic tumor on the right pretibial region that was initially considered to be a lipoma. Histopathology revealed the presence of a polymorphic inflammatory infiltrate within a myxoid and hyaline matrix. Interspersed between the inflammatory cells were 3 different populations of neoplastic cells: atypical spindle-shaped cells; bizarre epithelioid cells, some of which were multinucleated and resembled the virocytes or Reed-Sternberg cells; and cells with abundant, vacuolated cytoplasm, similar to lipoblasts. These clinical-pathologic findings led to a diagnosis of inflammatory myxohyaline tumor of the distal extremities. Although the tumor was excised with wide surgical margins, local recurrence developed after 3 months and was treated with re-excision and radiotherapy.
Los tumores subcutáneos suponen a menudo problemas significativos de diagnóstico diferencial en la clínica diaria que, al tiempo, condicionan dificultades en la decisión terapéutica. Presentamos el caso de un paciente con una masa subcutánea que resultó ser un tumor mixohialino inflamatorio de las zonas distales de las extremidades (TMHIADE), una neoplasia excepcional con un patrón histológico complejo y característico.
Caso clínicoMujer de 49 años que consultó por una masa subcutánea asintomática de 2 meses de evolución en la cara pretibial de la pierna derecha. No refería antecedentes de traumatismos ni cirugías locales en la zona de crecimiento del tumor. La masa había experimentado un crecimiento progresivo a lo largo de las últimas semanas generando discretas molestias. La exploración clínica mostró un nódulo cutáneo del mismo color de la piel, bien delimitado y de tacto firme y elástico, de 5 x 6 x 2cm, que planteó el diagnóstico de lipoma cutáneo o de tumor maligno de partes blandas (fig. 1). Los estudios analíticos mostraron normalidad en el recuento sanguíneo, electrolitos y bioquímica. El estudio radiológico permitió comprobar una tumoración subcutánea de tejidos blandos, no calcificada, que aparentemente no afectaba la fascia muscular subyacente. Se realizó una biopsia profunda tipo huso. El estudio histológico con hematoxilina-eosina mostró una masa tumoral dérmica y subcutánea, constituida por un infiltrado inflamatorio de poca densidad formado por linfocitos, neutrófilos, células plasmáticas, histiocitos y numerosos eosinófilos, inmerso en un estroma mixoide e hialino (fig. 2A). Entre este infiltrado inflamatorio existían tres poblaciones de células tumorales: por una parte, una proporción de las células que constituían la tumoración tenían un aspecto epitelioide, algunas de amplio citoplasma basófilo y núcleos bizarros hipercromáticos, con marcado pleomorfismo nuclear mientras que otras de estas células epitelioides eran multinucleadas, con núcleos vesiculares y nucléolos prominentes eosinofílicos semejantes a los virocitos o células de Reed-Sternberg (fig. 2B); la segunda población celular estaba constituida por unas células tumorales fusiformes de citoplasma basófilo y núcleos atípicos (fig. 2C). La tercera población celular estaba formada por unas células de citoplasma amplio multivacuoladas que recordaban a los lipoblastos (fig. 2 D). El estudio inmunohistoquímico mostró que las 3 poblaciones tumorales eran vimentina+++ (fig. 3A), con una positividad moderada para CD68 (fig. 3B) y CD34 (fig. 3C), y negativo para S-100, CD45 (antígeno leucocitario común), CD15, CD30, CD31, citoqueratina CAM 5.2, desmina y alfa-actina. Aunque la atipia celular era marcada, el número de mitosis fue moderado; el marcador de proliferación tumoral ki67 fue del 30% (fig. 3D). Teniendo en cuenta las características clínicas, histopatológicas e inmunohistoquímicas se llegó al diagnóstico de TMHIADE. Debido a este diagnóstico se realizó una extirpación tumoral amplia con márgenes de 2cm, con raspado del periostio y cierre con injerto cutáneo. La imagen macroscópica de la pieza de extirpación mostró una tumoración multinodular, con áreas hemorrágicas, que infiltraba la hipodermis alcanzando el periostio en forma de nódulos tumorales (fig. 1B). Los márgenes quirúrgicos fueron negativos para tumor, aunque el periostio resecado mostraba persistencia tumoral. La paciente experimentó una recidiva tumoral a los 3 meses de la intervención (fig. 4) que fue tratada con nueva extirpación quirúrgica e injerto y un ciclo de radioterapia postoperatoria. Actualmente, tras 9 meses de seguimiento la paciente está sana y libre de enfermedad.

A. Masa subcutánea mal delimitada en la zona pretibial derecha, de rápido crecimiento en las últimas semanas, inicialmente diagnosticada de lipoma.
B. Imagen macroscópica: tumor multinodular subcutáneo mal delimitado de 5x6,5x2cm, de aspecto gelatinoso y presencia de áreas hemorrágicas, que infiltra tejido adiposo y la grasa perióstica en forma de nódulos tumorales.

A. Hematoxilina-eosina x200: focos de infiltrado inflamatorio polimórfico denso, con presencia de linfocitos, células plasmáticas, neutrófilos y numerosos eosinófilos, inmerso en la matriz mixoide, formando parte del tumor. B. Hematoxilina-eosina x400: población tumoral constituida por células tumorales multinucleadas a tipo Reed-Sternberg, y células epitelioideas.
C. Hematoxilina-eosina x400: células tumorales de morfología fusiforme.
D. Hematoxilina-eosina x400: células multivacuoladas de tipo lipoblasto.

Tinciones inmunohistoquímicas. A. Marcada positividad de las células tumorales para vimentina. B. Positividad moderada de las células tumorales para CD68. C. Positividad de las células tumorales para CD34. D. Índice de proliferación tumoral con el marcador Ki67 que mostraba una positividad del 20-30%.

El tumor mixohialino inflamatorio de las áreas distales de las extremidades (TMHIADE), también denominado sarcoma fibroblástico mixoinflamatorio acral, o tumor mixohialino inflamatorio distal de las extremidades con virocitos o células a tipo Reed-Sternberg, fue descrito por primera vez por Montgomery en 19971.
El TMHIADE es un sarcoma de bajo grado de malignidad, extremadamente infrecuente, con tendencia a las recurrencias locales, sobre todo tras una extirpación quirúrgica incompleta, pero con un bajo potencial metastásico1,2. Afecta por igual a ambos sexos, con un pico de incidencia en la cuarta y quinta décadas de la vida. La mayoría de pacientes comienzan con una masa generalmente indolora, mal delimitada, de crecimiento progresivo en los miembros y áreas distales3. El tiempo medio de evolución es de un año, aunque existen casos de rápido crecimiento en escasas semanas. Los miembros superiores se afectan con mayor frecuencia que los inferiores: los dedos y las manos son las localizaciones más frecuentes, seguidas de los brazos. En los miembros inferiores los dedos de los pies y el dorso del pie son las localizaciones más frecuentes, seguidas de las zonas distales de las piernas. Algunos pacientes refieren molestias locales o leve incapacidad funcional, sobre todo en los casos de rápido crecimiento tumoral3,4. El diagnóstico clínico previo a la biopsia suele ser de lesión benigna (60%)1 de tipo inflamatorio como tenosinovitis, o tipo tumoral, siendo el lipoma uno de los diagnósticos clínicos más frecuente, como ocurrió en nuestro caso. La resonancia magnética nuclear es muy útil en el diagnóstico al proporcionar hallazgos compatibles con un tumor invasivo de partes blandas, en forma de una masa subcutánea mal delimitada que se extiende al tejido adiposo subyacente y a los tendones, en caso de localizarse sobre ellos.
El TMHIADE es un tumor multinodular y mal circunscrito, por lo que normalmente la extirpación inicial suele ser parcial. Su tamaño medio es de 4cm (1-9cm)3.
Macroscópicamente presenta áreas de aspecto gelatinoso, por la abundante presencia de mucina, y áreas hemorrágicas, hallazgos que observamos en nuestro caso. En un porcentaje significativo de casos se asocia con sinovitis. El estudio histológico tiene como hallazgo más característico la presencia de un importante componente inflamatorio que se extiende de forma difusa por las áreas mixoides e hialinas del tumor, constituido por neutrófilos, linfocitos, células plasmáticas y eosinófilos, entremezcladas con una trama vascular muy rica, que simula un tejido de granulación4. La proporción de áreas mixoides e hialinas varía de unos casos a otros, e incluso dentro de una misma tumoración. En algunos casos la tumoración está mayoritariamente constituida por áreas hipocelulares mixoides, mientras que en otros los depósitos de mucina son más focales5. En las zonas mixo-hialinas las células tumorales se entremezclan con células inflamatorias. En las zonas de mayor densidad tumoral las células neoplásicas se dividen en 3 poblaciones de diferente morfología: el primer tipo celular son células fusiformes bizarras, de citoplasma basófilo y núcleos atípicos, como se observó en nuestro caso. El segundo tipo son células epitelioides, grandes, poligonales, de tipo ganglión, de citoplasma basófilo y núcleo oval, con llamativa atipia nuclear, cromatina vesicular y nucléolos prominentes de gran tamaño1, algunas de ellas multinucleadas similares a los virocitos o células Reed-Sternberg que se observan en la enfermedad de Hodgkin, y que también existían en nuestro paciente. En ocasiones, estas células gigantes presentan linfocitos, eosinófilos y neutrófilos en su interior (incluso células plasmáticas y hematíes) produciendo fenómenos de emperipolesis, que no observamos en nuestro caso1,6–8. El tercer tipo está representado por células multivacuoladas de tamaños variables, con un amplio citoplasma eosinófilo, semejantes a los lipoblastos pleomórficos, y que también detectamos en el estudio histológico de nuestro caso. Las mitosis no suelen superar la cifra de 1 por campo de gran aumento, con un índice proliferativo < 5% en > 90% de los casos1, y no existen figuras mitóticas atípicas. Tampoco suele observarse necrosis en este tipo de tumor. La inmunohistoquímica del TMHI muestra que las células tumorales son difusamente positivas para vimentina, con células multinucleadas dispersas CD68+. Algunas de las células pueden mostrar positividad para CD34. El resto de marcadores, como citoqueratina CAM 5.2, queratina AE1:AE3, antígeno epitelial de membrana (EMA), proteína S100, HMB-45, Melan-A, actina de músculo liso, desmina, factor VIII, CD15, CD30, antígeno leucocitario común (LCA) y CD15 (Leu-M1) son negativos1,6,8-9.
El diagnóstico del TMHIADE no es sencillo y presenta unas dificultades inherentes a las particularidades histopatológicas de este sarcoma. El intento diagnóstico mediante una biopsia superficial puede generar errores diagnósticos, ya que esta tumoración podría ser confundida fácilmente con un tumor mixoide benigno o un proceso inflamatorio. De hecho, antes de su descripción definitiva en 1997, este sarcoma solía diagnosticarse erróneamente de enfermedad de Hodgkin, de sarcomas de tejidos blandos sin otra especificación o incluso de procesos inflamatorios o infecciosos1,2,4. Esto último se debe a que la mayor peculiaridad histopatológica del TMHIADE es la presencia de un infiltrado inflamatorio polimorfo, en la mayoría de casos muy abundante, que también ha sido descrito en otros tipos de sarcomas, pero que es especialmente relevante en este tipo: en ocasiones, las células tumorales son escasas y de distribución dispersa, por lo que pueden quedar enmascaradas dentro del denso infiltrado inflamatorio, estableciéndose de forma equívoca el diagnóstico de proceso reactivo o pseudotumor inflamatorio1-3,5. Además, una biopsia superficial no nos permitirá observar la infiltración del tejido adiposo por las células tumorales, que pueden alcanzar los tendones y el periostio, circunstancias muy frecuentes en el TMHIADE e infrecuentes en los procesos inflamatorios. Por ello, siempre es recomendable realizar una biopsia profunda ante la sospecha de un tumor de partes blandas. Una de las entidades que habría que considerar en el diagnóstico diferencial del TMHIADE con los procesos reactivos es la enfermedad de Rosai-Dorfman, que también presenta fenómenos de emperipolesis, aunque sin vacuolas citoplasmáticas ni intranucleares, que sí presentan las células tumorales del TMHI, y con una inmunohistoquímica positiva para S-100. El TMHIADE es negativo para este marcador1,2,8.
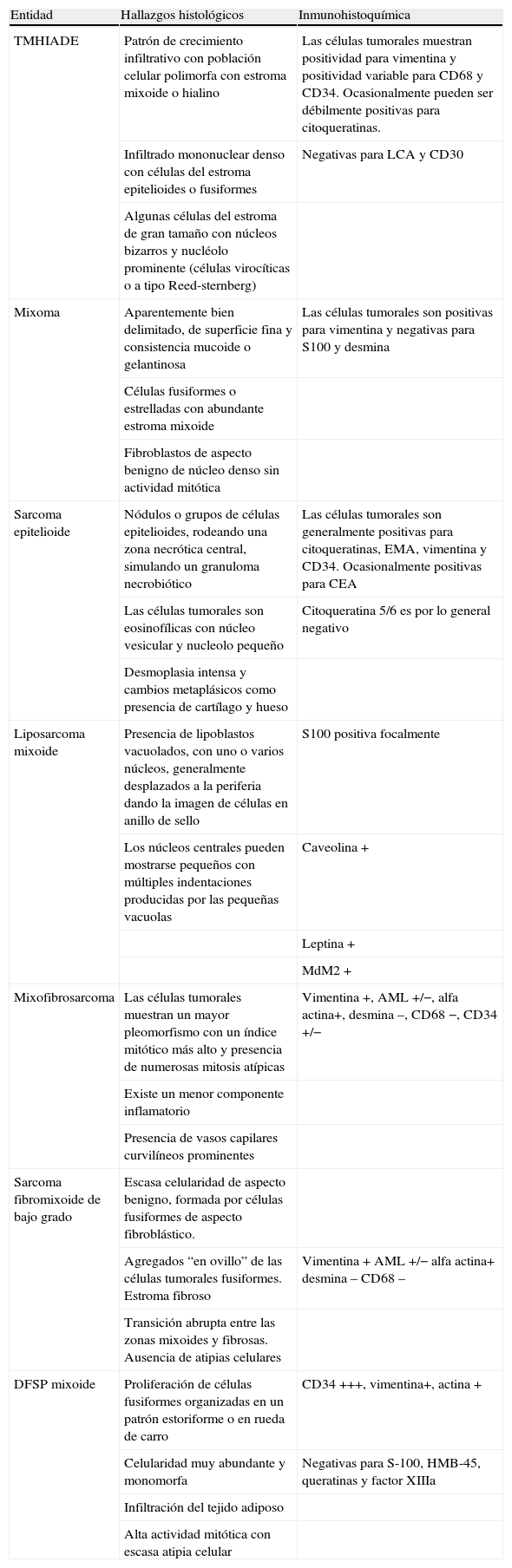
El diagnóstico diferencial del TMHIADE es amplio y dependerá del componente que predomine en el tumor: mixoide, inflamatorio o células bizarras atípicas. En primer lugar, habría que diferenciarlos de otros tumores de tejidos blandos de tipo mixoide, benignos y malignos, fundamentalmente con la variante mixoide del histiocitoma fibroso maligno (mixofibrosarcoma) de bajo grado. Este tumor suele afectar a individuos de edad más avanzada (sexta a octava décadas) y normalmente aparece en las áreas proximales de las extremidades, a diferencia de la localización distal del TMHIADE. Respecto a los hallazgos histológicos la celularidad es más pleomórfica, el índice mitótico es mucho mayor en el mixofibrosarcoma que en el TMHIADE, con una alta tasa de mitosis atípicas9. Además, estos tumores pueden presentar una reacción inflamatoria asociada, pero carecen de fenómenos de emperipolesis y no suele ser tan llamativa como el TMHIADE. El estudio inmunohistoquímico del mixofibrosarcoma revela que las células son vimentina +, algunas células expresan actina muscular lisa y alfa actina+ y, al contrario de lo que ocurre en el TMHIADE, suelen ser negativas para desmina, CD68 y CD34. El mixofibrosarcoma implica peor pronóstico que el TMHIADE, ya que presenta una mayor tasa de metástasis a distancia (tabla 1)1-4,8,9. Otros tumores malignos con estroma mixoide con los que habría que establecer el diagnóstico diferencial son: el liposarcoma mixoide, sarcoma fibromixoide de bajo grado, tumor de la vaina tendinosa de los nervios periféricos maligno mixoide, la variante mixoide del dermatofibrosarcoma protuberans (tabla 1) y el condrosarcoma mixoide extraesquelético.
Diagnóstico diferencial del tumor mixohialino inflamatorio de las áreas distales de las extremidades (TMHIADE) que incluye tumores mixoides benignos y malignos, como el mixoma, mixofibrosarcoma, sarcoma fibromixoide de bajo grado, DFSP mixoide y liposarcoma mixoide y sarcomas de localización distal, como el sarcoma epiteloide.
| Entidad | Hallazgos histológicos | Inmunohistoquímica |
| TMHIADE | Patrón de crecimiento infiltrativo con población celular polimorfa con estroma mixoide o hialino | Las células tumorales muestran positividad para vimentina y positividad variable para CD68 y CD34. Ocasionalmente pueden ser débilmente positivas para citoqueratinas. |
| Infiltrado mononuclear denso con células del estroma epitelioides o fusiformes | Negativas para LCA y CD30 | |
| Algunas células del estroma de gran tamaño con núcleos bizarros y nucléolo prominente (células virocíticas o a tipo Reed-sternberg) | ||
| Mixoma | Aparentemente bien delimitado, de superficie fina y consistencia mucoide o gelantinosa | Las células tumorales son positivas para vimentina y negativas para S100 y desmina |
| Células fusiformes o estrelladas con abundante estroma mixoide | ||
| Fibroblastos de aspecto benigno de núcleo denso sin actividad mitótica | ||
| Sarcoma epitelioide | Nódulos o grupos de células epitelioides, rodeando una zona necrótica central, simulando un granuloma necrobiótico | Las células tumorales son generalmente positivas para citoqueratinas, EMA, vimentina y CD34. Ocasionalmente positivas para CEA |
| Las células tumorales son eosinofílicas con núcleo vesicular y nucleolo pequeño | Citoqueratina 5/6 es por lo general negativo | |
| Desmoplasia intensa y cambios metaplásicos como presencia de cartílago y hueso | ||
| Liposarcoma mixoide | Presencia de lipoblastos vacuolados, con uno o varios núcleos, generalmente desplazados a la periferia dando la imagen de células en anillo de sello | S100 positiva focalmente |
| Los núcleos centrales pueden mostrarse pequeños con múltiples indentaciones producidas por las pequeñas vacuolas | Caveolina + | |
| Leptina + | ||
| MdM2 + | ||
| Mixofibrosarcoma | Las células tumorales muestran un mayor pleomorfismo con un índice mitótico más alto y presencia de numerosas mitosis atípicas | Vimentina +, AML +/−, alfa actina+, desmina –, CD68 −, CD34 +/− |
| Existe un menor componente inflamatorio | ||
| Presencia de vasos capilares curvilíneos prominentes | ||
| Sarcoma fibromixoide de bajo grado | Escasa celularidad de aspecto benigno, formada por células fusiformes de aspecto fibroblástico. | |
| Agregados “en ovillo” de las células tumorales fusiformes. Estroma fibroso | Vimentina + AML +/− alfa actina+ desmina – CD68 – | |
| Transición abrupta entre las zonas mixoides y fibrosas. Ausencia de atipias celulares | ||
| DFSP mixoide | Proliferación de células fusiformes organizadas en un patrón estoriforme o en rueda de carro | CD34 +++, vimentina+, actina + |
| Celularidad muy abundante y monomorfa | Negativas para S-100, HMB-45, queratinas y factor XIIIa | |
| Infiltración del tejido adiposo | ||
| Alta actividad mitótica con escasa atipia celular |
AML: actina muscular lisa; DFSP: dermatofibrosarcoma protuberans; EMA: antígeno epitelial de membrana; LCA: antígeno leucocitario común; S-100: proteína S-100; TMHIADE: tumor mixohialino inflamatorio de las áreas distales de las extremidades.
Otro proceso que debe ser incluido en el diagnóstico diferencial es el sarcoma epitelioide, que también aparece en pacientes jóvenes y de mediana edad y en áreas distales. La presencia de células de tipo Reed-Sternberg plantea el diagnóstico diferencial con la enfermedad de Hodgkin, aunque la presencia del resto de poblaciones tumorales características del TMHIADE descartaría este diagnóstico.
También habría que diferenciarlo de diversos tumores benignos de tejidos blandos, de entre los cuales destaca el mixoma (tabla 1). Este diagnóstico puede llegar a plantearse sobre todo en los casos de TMHIADE con escasa celularidad y con un componente mixoide muy marcado, en los que se realiza una biopsia superficial que no nos permite detectar la celularidad maligna del TMHIADE. Otros tumores benignos con los que habría que establecer el diagnóstico diferencial son: lipoma mixoide, quiste ganglión y neurofibroma mixoide.
Los estudios ultraestructurales han permitido conocer que las células tumorales del TMHIADE tienen características de fibroblastos modificados, incluyendo abundantes filamentos intermedios y un retículo endoplásmico rugoso dilatado. No se han podido demostrar anomalías citogenéticas asociadas de una manera constante a este sarcoma: se han descrito translocación t(1;10)10 y cromosomas dicéntricos secundarios a la rotura del cromosoma 7 y 8,6 translocación t(2,6)11 y pérdidas en los cromosomas 3 y 1312, entre otros.
El TMHIADE tiene una gran tendencia a la recidiva local, sobre todo si la extirpación quirúrgica resulta incompleta, como ocurrió en nuestro caso. Estas recidivas muchas veces (> 50%) son múltiples y agresivas, y requieren la amputación del miembro hasta en un 30% de los casos. Las metástasis a distancia se han descrito ocasionalmente a ganglios linfáticos distantes y a los pulmones, con una tasa del 2-6% a los 5 años. El tratamiento de elección es una extirpación quirúrgica amplia, consiguiendo unos márgenes negativos para el tumor. En caso de recidiva se aconseja la reintervención quirúrgica. Como tratamientos adyuvantes, en casos irresecables o recidivantes, se podría plantear el tratamiento con radioterapia o inhibidores del EGFR, especialmente en aquellos tumores con células positivas para EGFR7,9,13.
En definitiva, presentamos un caso de TMHIADE, entidad infrecuente que debe conocerse tanto por su frecuente confusión clínica con entidades benignas, como por las dificultades y amplio diagnóstico diferencial que supone su correcto diagnóstico histopatológico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



