






La familia del tumor neuroectodérmico primitivo periférico/sarcoma de Ewing (PNET/ES) representa a un grupo de neoplasias malignas poco frecuentes, incluidas dentro de un grupo de tumores que presentan característicamente en su histología células redondas de pequeño tamaño. La traslocación 11;22 es típica de este tumor. Se presenta el caso de un varón de 45 años con un tumor subcutáneo neuroectodérmico primitivo periférico, en el que se detectó la traslocación t(11;22), tratado satisfactoriamente mediante cirugía y radioterapia.
Peripheral primitive neuroectodermal tumors—also known as Ewing sarcomas—are a rare type of malignant tumor the histology of which characteristically reveals the presence of small round cells. Typically, t(11;22) translocation is observed. We describe the case of a 45-year-old man with a subcutaneous peripheral primitive neuroectodermal tumor in which the t(11;22) translocation was detected. He was satisfactorily treated with surgery and radiotherapy.
Un paciente varón de 45 años, sin antecedentes personales ni familiares de interés, consultó para valorar la presencia de una lesión asintomática en el hombro derecho de crecimiento progresivo, y que presentaba desde hacía cinco meses. Negaba haber sufrido pérdida de peso, astenia, fiebre o malestar general durante ese período de tiempo.
En la exploración física se observaba una lesión tumoral subcutánea de 6 × 7cm de diámetro, eritemato-violácea, con costras y telangiectasias en superficie, de consistencia firme y adherida a planos profundos (fig. 1), sin palparse adenopatías locorregionales. Tanto el hemograma, como la bioquímica rutinaria practicados, no presentaron alteraciones significativas. En el estudio ecográfico se apreciaba una lesión de 3,8 × 4,3 × 3,8cm con contornos nítidos y lisos, y con un contenido irregular a modo de mamelones hiperecogénicos, vasos sanguíneos, así como zonas quísticas de contenido líquido. Se realizó una punción aspiración con aguja fina (PAAF), extrayéndose material de aspecto achocolatado que se envió al laboratorio de anatomía patológica, cuyo estudio describió una población de células uniformes, desprovistas de citoplasma, núcleos de cromatina irregular y nucleolo ausente o poco prominente, que sugerían un tumor maligno de células redondas, posiblemente de estirpe neuroectodérmica.

El examen histopatológico de la biopsia mostró una proliferación neoplásica localizada en la hipodermis, que se extendía hacia la dermis profunda respetando la epidermis, y constituida por células redondas de mediano tamaño. Estas exhibían un núcleo redondo u oval con cromatina finamente granular, nucleolo poco prominente y escaso citoplasma (fig. 2), así como pequeñas vacuolas intracitoplasmáticas. Las células se disponían formando masas o nidos sólidos y difusos, con una rica vascularización y escaso estroma. En la tumoración se advertían diversos patrones, destacando zonas de disposición alveolar, masas lobuladas, áreas angiomatoides o pseudoglandulares. En algunas zonas se insinuaban imágenes de tipo pseudo-roseta (fig. 3). Las mitosis eran escasas, apreciándose asimismo pequeños focos de hemorragia con depósitos hemosiderínicos. La inmunohistoquimia reveló positividad para enolasa neuronal específica y negatividad para cromogranina A, vimentina, sinaptofisina, citoqueratinas 7 y 20, antígeno epitelial de membrana, HMB-45, proteína S-100, CD43HT1, AE1-AE y CD45RBLCA. La tinción con CD 99 no fue realizada.
.")
.")
El análisis citogenético de la muestra, realizado mediante hibridación fluorescente in situ (FISH), detectó la rotura del gen EWS resultante de la traslocación t(11;22) que, junto con los hallazgos anteriores, confirmó el diagnóstico de tumor subcutáneo neuroectodérmico primitivo periférico (fig. 4).
 detectada mediante fluorescencia hibridación fluorescente in situ (FISH).")
En la resonancia magnética nuclear (RMN) practicada se apreciaba una masa subcutánea bien delimitada, localizada en el tejido celular subcutáneo, que conectaba con la fascia del músculo deltoides sin infiltrarlo. La tomografía axial computarizada (TAC) no mostró la presencia de enfermedad metastásica, con lo cual se realizó la extirpación de la lesión usando 2cm de margen, incluyendo la fascia muscular y cubriendo el defecto mediante un injerto de piel total (fig. 5).

Como terapia complementaria, se realizó un tratamiento radioterápico del lecho tumoral mediante acelerador lineal de electrones (6 Mev), con una dosis total de 66Gy con 2Gy/fracción/día, 5 días a la semana. Igualmente se trataron las cadenas ganglionares de axila derecha con 50Gy (2Gy/fracción y día, 5 días a la semana) con fotones. Posteriormente en el servicio de Oncología Médica se añadió un esquema terapéutico con vincristina 2mg, adriamicina 75mg/m2 y ciclofosfamida 1.200mg/m2 durante 1 año, añadiendo cada 21 días factor estimulante de colonias granulocíticas.
Tras 25 meses de seguimiento, el paciente continúa libre de enfermedad.
DiscusiónBajo la denominación de tumores de células redondas pequeñas, se agrupan una serie de neoplasias malignas que muestran en su histología una proliferación de elementos celulares monomorfos e indiferenciados, con un elevado cociente entre núcleo y citoplasma1. Muchos de ellos presentan características clinicopatológicas superponibles, siendo necesario estudios inmunohistoquímicos, de microscopía electrónica y citogenéticos para alcanzar un diagnóstico correcto. La baja frecuencia de estos tumores, así como la escasez de casos publicados, hacen difícil extrapolar datos que ayuden a conocer el pronóstico o el tipo de tratamiento más adecuado.
El término tumor neuroectodérmico primitivo alude a una serie de neoplasias con diferenciación hacia tejidos neuronales. Es por ello que pueden afectar al sistema nervioso central, a los ganglios del sistema autosómico parasimpático, así como a los tejidos blandos, pudiendo interesar a los nervios periféricos. Es en este último caso donde se adopta la denominación de tumor neuroectodérmico periférico primitivo (PNET)2. Gran parte de los autores consideran al sarcoma de Ewing (ES) como la misma entidad que el PNET, dado que comparten la traslocación t(11;22). Sin embargo, otros autores lo separan, puesto que el ES no presenta gránulos neurosecretores, siendo considerado como una forma más primitiva3.
La forma cutánea del PNET/ES es aun menos frecuente, habiendo sido publicados hasta la fecha menos de 30 casos. Aunque predomina en niños o en la etapa adolescente, la edad de presentación puede ser muy variable, oscilando el rango desde los 22 meses hasta los 81 años. No existe localización preferente para este tumor, ya que se han descrito en el tronco, las extremidades, la cara o el cuero cabelludo. Tampoco se han descrito diferencias en cuanto al sexo o la raza4.
No se han detectado factores ambientales desencadenantes, ni tampoco agregación familiar.
Clínicamente se desarrolla como nódulos o masas cutáneas o subcutáneas, de consistencia firme o elástica, de crecimiento lento y discretamente doloroso. Su coloración a menudo es similar a la de la piel sana, o adoptan una tonalidad eritemato-violácea4,5.
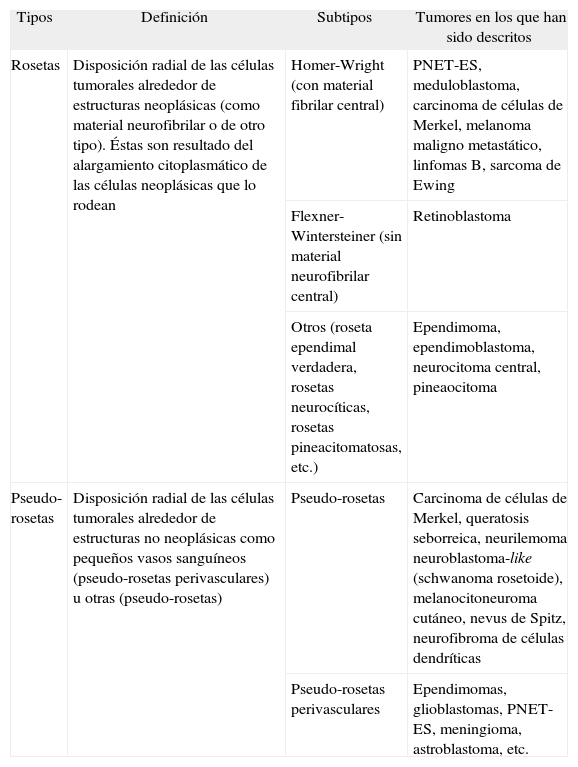
Histológicamente el PNET/ES puede asentar en la dermis profunda o en el tejido celular subcutáneo. Se caracteriza por proliferaciones densas y sólidas de células redondas pequeñas, que en ocasiones se disponen en cordones, nidos o lóbulos. Sin embargo, en un 10-20 % de los casos se encuentran células con aspecto fusiforme6. Es frecuente encontrar espacios microquísticos o pseudovasculares que pueden adoptar un patrón retiforme. También es frecuente el hallazgo de material neurófilo y rosetas de tipo Homer-Wright (tabla 1). Los tumores suelen tener septos fibro-vasculares y áreas focales de necrosis. Individualmente, las células neoplásicas presentan un escaso citoplasma eosinófilo con núcleo hipercromático oval y cromatina dispersa, y suele encontrarse, a diferencia de nuestro caso, intensa actividad mitótica. En algunos casos publicados se han identificado células rabdoides y plasmocitoides4.
Tipos de rosetas y pseudo-rosetas
| Tipos | Definición | Subtipos | Tumores en los que han sido descritos |
| Rosetas | Disposición radial de las células tumorales alrededor de estructuras neoplásicas (como material neurofibrilar o de otro tipo). Éstas son resultado del alargamiento citoplasmático de las células neoplásicas que lo rodean | Homer-Wright (con material fibrilar central) | PNET-ES, meduloblastoma, carcinoma de células de Merkel, melanoma maligno metastático, linfomas B, sarcoma de Ewing |
| Flexner-Wintersteiner (sin material neurofibrilar central) | Retinoblastoma | ||
| Otros (roseta ependimal verdadera, rosetas neurocíticas, rosetas pineacitomatosas, etc.) | Ependimoma, ependimoblastoma, neurocitoma central, pineaocitoma | ||
| Pseudo-rosetas | Disposición radial de las células tumorales alrededor de estructuras no neoplásicas como pequeños vasos sanguíneos (pseudo-rosetas perivasculares) u otras (pseudo-rosetas) | Pseudo-rosetas | Carcinoma de células de Merkel, queratosis seborreica, neurilemoma neuroblastoma-like (schwanoma rosetoide), melanocitoneuroma cutáneo, nevus de Spitz, neurofibroma de células dendríticas |
| Pseudo-rosetas perivasculares | Ependimomas, glioblastomas, PNET-ES, meningioma, astroblastoma, etc. |
ES: sarcoma de Ewing; PNET: tumor neuroectodérmico primitivo periférico.
Ultraestructuralmente se suelen observar células redondas con escaso citoplasma y grandes núcleos ovoides. Estos núcleos muestran heterocromatina densa y pequeños nucleolos. En el citoplasma es posible encontrar microtúbulos, gránulos neuroendocrinos y filamentos intermedios7,8.
Los estudios inmunohistoquímicos pueden exhibir positividad para vimentina, β2-microglobulina, CD 99 (antígeno de membrana MIC 2), así como para otros marcadores neuronales (proteína S-100, Leu-7, neurofilamentos, enolasa neuronal específica, sinaptofisina y PGP9.5). Estos marcadores son de gran importancia para establecer el diagnóstico diferencial con otras entidades9.
La detección mediante FISH o por reacción en cadena de la polimerasa con transcripción inversa (PCR-RT) de la traslocación, tanto de t(11;22)(q24;q12) como de t(21;22) (q2;q12), se considera diagnóstica de PNET/ES10,11. En concreto, la traslocación t(11;22)(q24;q12) produce la fusión del gen EWS (localizado en 22q12) con el gen Fli1 (que se sitúa en 11q24). Fli1 pertenece a una familia de factores de transcripción de un grupo de proteínas llamadas «Ets». Esta fusión generaría un factor de transcripción aberrante que promovería la tumorogénesis. Ambas traslocaciones se detectan en el 90-95 % de los casos. Recientemente se ha descubierto una tercera traslocación –t(7;22)–que se ha relacionado con un peor pronóstico, al desarrollar una enfermedad más agresiva12,13.
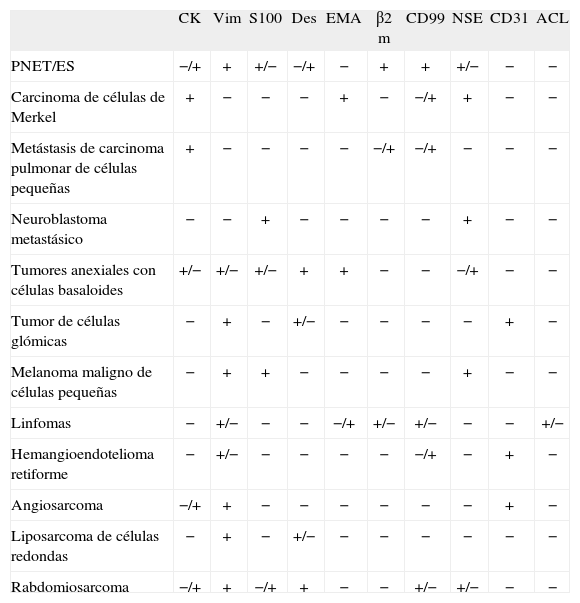
El diagnóstico diferencial se plantea fundamentalmente con los tumores de células redondas, e incluye el carcinoma de células de Merkel14, el carcinoma pulmonar metastático de células pequeñas, el neuroblastoma metastático, las neoplasias anexiales cutáneas basaloides, el melanoma maligno de células pequeñas, los linfomas, el hemangioendotelioma retiforme, el angiosarcoma, el liposarcoma de células redondas y el rabdomiosarcoma, entre otros. Como ya hemos señalado, la inmunohistoquímica desempeña aquí un papel clave para llegar a un diagnóstico acertado15 (tabla 2).
Diagnóstico diferencial inmunohistoquímico de los tumores que presentan células redondas en su histología
| CK | Vim | S100 | Des | EMA | β2m | CD99 | NSE | CD31 | ACL | |
| PNET/ES | −/+ | + | +/− | −/+ | − | + | + | +/− | − | − |
| Carcinoma de células de Merkel | + | − | − | − | + | − | −/+ | + | − | − |
| Metástasis de carcinoma pulmonar de células pequeñas | + | − | − | − | − | −/+ | −/+ | − | − | − |
| Neuroblastoma metastásico | − | − | + | − | − | − | − | + | − | − |
| Tumores anexiales con células basaloides | +/− | +/− | +/− | + | + | − | − | −/+ | − | − |
| Tumor de células glómicas | − | + | − | +/− | − | − | − | − | + | − |
| Melanoma maligno de células pequeñas | − | + | + | − | − | − | − | + | − | − |
| Linfomas | − | +/− | − | − | −/+ | +/− | +/− | − | − | +/− |
| Hemangioendotelioma retiforme | − | +/− | − | − | − | − | −/+ | − | + | − |
| Angiosarcoma | −/+ | + | − | − | − | − | − | − | + | − |
| Liposarcoma de células redondas | − | + | − | +/− | − | − | − | − | − | − |
| Rabdomiosarcoma | −/+ | + | −/+ | + | − | − | +/− | +/− | − | − |
ACL: antígeno leucocitario común; β2m: β-2 microglobulina; CK: citoqueratina; Des: desmina; EMA: antígeno epitelial de membrana; NSE: enolasa neuronal específica; Vim: vimentina.
+: positivo > 90 % casos; +/−: ocasionalmente positivo; −/+: positivo en casos aislados; -: negativo.
Debido a la escasez de casos publicados, no conocemos con exactitud la historia natural del PNET/ES, aunque se postula que pueda tener un comportamiento agresivo en aquellos casos que se desarrollan en zonas profundas, cuando son tumores de gran tamaño o presenten la traslocación t(7;22)12.
Taylor8 describe una serie de 31 pacientes afectos de PNET/ES, de los cuales cuatro fallecieron de enfermedad metastásica, 5 desarrollaron metástasis ganglionares locorregionales y los restantes casos permanecieron libres de enfermedad transcurridos 42 meses. No parece haber correlación entre el tipo clínico-histopatológico y la capacidad metastásica, pero sí con la presencia intensa de necrosis o de invasión vascular.
Por la misma razón, no sabemos cuál es el tratamiento más adecuado para este tumor, pero generalmente se acepta que la combinación de cirugía, radioterapia y quimioterapia consigue un aumento notable de la supervivencia y del tiempo libre de enfermedad.
ConclusiónPresentamos un caso de PNET/ES subcutáneo, con hallazgos citogenéticos típicos, que ha respondido satisfactoriamente al tratamiento combinado con cirugía, radioterapia y quimioterapia. El amplio diagnóstico diferencial que plantea hace de la histología, la inmunohistoquímica y la citogenética herramientas imprescindibles para llegar a un diagnóstico correcto.
Conflicto de interesesDeclaramos no tener ningún conflicto de intereses.



