


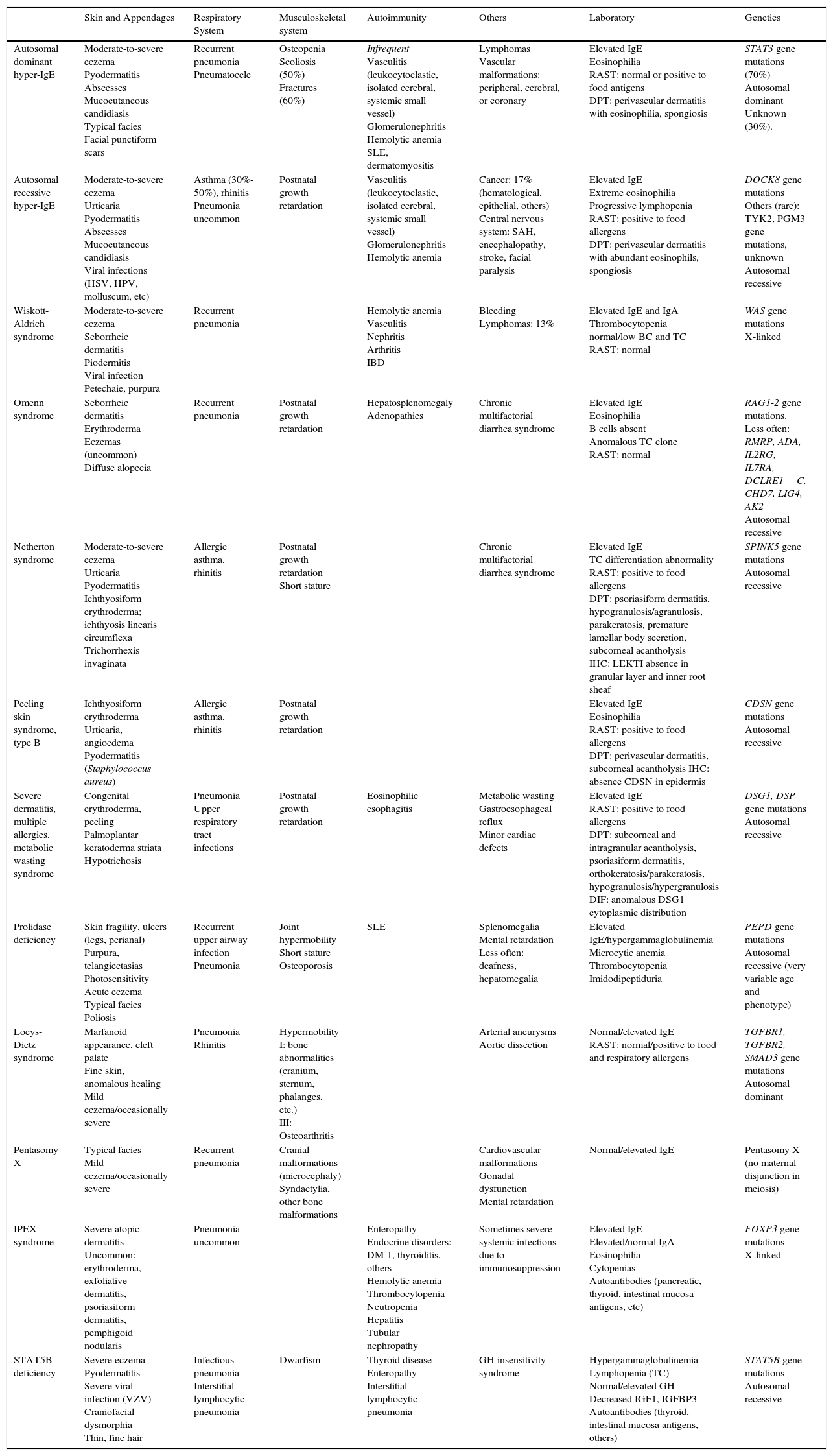
The association of moderate to severe eczema and elevated plasma levels of immunoglobulin E is a characteristic not only of atopic dermatitis but also of various genodermatoses: hyperimmunoglobulin E syndromes, Omenn syndrome, Netherton syndrome, peeling skin syndrome type B, severe dermatitis, multiple allergies, and metabolic wasting syndrome, Wiskott-Aldrich syndrome, prolidase deficiency, Loeys-Dietz syndrome, IPEX syndrome, STAT5B deficiency, and pentasomy X. The clinical presentation of these genodermatoses –typically in children– is consistent with severe atopic dermatitis. Immunoglobulin E is elevated from birth and response to conventional treatments is poor. Diagnosis is further complicated by the fact that these genodermatoses often share other clinical manifestations and laboratory findings. We present practical guidelines for differentiating among these various entities, with the aim of helping physicians decide what type of genetic test should be carried out –and when– in order to establish a definitive diagnosis.
La asociación de eccema moderado-grave y niveles elevados de IgE en plasma es característica no solo de la dermatitis atópica, sino también de diversas genodermatosis: síndromes hiper-IgE, síndrome de Omenn, síndrome de Netherton, síndrome de la piel exfoliada tipo B, síndrome de dermatitis grave-alergias múltiples-desgaste metabólico, síndrome de Wiskott-Aldrich, déficit de prolidasa, síndrome de Loeys-Dietz, síndrome IPEX, déficit de STAT5B y pentasomía X. Se trata de pacientes pediátricos que presentan un cuadro clínico compatible con dermatitis atópica grave, con mala respuesta a los tratamientos clásicos y que asocian elevación de IgE desde el nacimiento. Además, comparten con frecuencia otras manifestaciones clínicas y analíticas, lo cual dificulta el diagnóstico. Presentamos una guía práctica para orientar el diagnóstico diferencial entre todas estas entidades y, por lo tanto, ayudar a decidir cuándo y el tipo de test genético a realizar para establecer el diagnóstico definitivo.
The association between moderate-to-severe eczema and elevated plasma immunoglobulin (Ig) E levels is characteristic of atopic dermatitis and is also seen in other genetic diseases. Many patients also have concurrent congenital immunodeficiency. When faced with a patient with clinical signs and symptoms consistent with severe atopic dermatitis1 who does not respond to traditional treatments (hygiene and dietary interventions, topical or systemic corticosteroids, cyclosporin, methotrexate, or azathioprine)1 and has elevated IgE from the first days of life, we should suspect the presence of one of these syndromes.
One group, hyper-IgE syndromes, has been described and extensively studied. These entities are classified according to the pattern of transmission: autosomal dominant (AD) or autosomal recessive (AR). Both types are characterized by elevated IgE and a clinical picture dominated by skin involvement, with moderate or severe acute and subacute eczema that resembles atopic dermatitis, and by recurrent skin and respiratory infection.
The other group comprises a series of low-prevalence congenital diseases that often present with episodes of refractory eczema associated with elevated IgE and that should be included in our diagnostic algorithm. Omenn syndrome is a serious combined immunodeficiency of AR transmission that presents in the first year of life. Wiskott-Aldrich syndrome is a primary X-linked immunodeficiency characterized essentially by bleeding diathesis secondary to thrombocytopenia or platelet dysfunction, along with recurrent eczemas and bacterial infections from birth. Prolidase deficiency is a multisystemic disease of AR transmission with a very variable clinical presentation and severity, associated with limited or no prolidase enzyme in erythrocytes, leukocytes, or fibroblasts. Loeys-Dietz syndrome is a connective tissue disease of AD transmission that is associated with a marfanoid appearance and abnormalities in the great arteries. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a serious congenital autoimmune disease in which refractory diarrhea, infections, and multiple endocrine disorders are present in addition to skin involvement. STAT5B deficiency is a condition of AR transmission included in the group of syndromes related to growth hormone insensitivity. This entity is notable for the occurrence of primary immunodeficiency. Pentasomy X is a congenital disease caused by chromosomal abnormalities in women leading to gonadal dysfunction, delayed development, short stature, and musculoskeletal and craniofacial malformations. Netherton syndrome is an AR genodermatosis characterized by ichthyosiform erythroderma, trichorrhexis invaginata, and atopic manifestations present almost from birth. Finally, we include 2 very recently described genodermatosis related to Netherton syndrome from the pathophysiological point of view: peeling skin syndrome type B (PSS-B) and severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome. PSS-B is an AR genodermatosis with ichthyosiform erythroderma from birth which, in addition, presents with severe food allergies, angioedema, and urticaria. Finally, SAM is another rare AR genodermatosis in which skin involvement is combined with notable food allergies, esophageal involvement, and other characteristics that will be discussed later in this article.
Differential diagnosis of these syndromes can be very complex, given that there may be considerable overlap and many clinical characteristics are shared. The phenotypic expression of each of these conditions may also vary from one individual to another. Thus, in this article, we provide key information to enable differential diagnosis from the clinical point of view, with the intention of guiding which genetic studies to request. Definitive diagnosis is made by identifying the causal mutation. In Table 1, we have compiled the main genodermatosis that should be included in the differential diagnosis, indicating the main characteristics to investigate in the diagnostic procedure to enable us reach a full diagnosis. A complete detailed description of each of these diseases is not an objective of this article. For this, the reader is referred to literature for more information.
Differential Clinical and Laboratory Diagnosis of Genodermatoses That Present With Moderate-to-Severe Eczema and Elevated IgE.
| Skin and Appendages | Respiratory System | Musculoskeletal system | Autoimmunity | Others | Laboratory | Genetics | |
|---|---|---|---|---|---|---|---|
| Autosomal dominant hyper-IgE | Moderate-to-severe eczema Pyodermatitis Abscesses Mucocutaneous candidiasis Typical facies Facial punctiform scars | Recurrent pneumonia Pneumatocele | Osteopenia Scoliosis (50%) Fractures (60%) | Infrequent Vasculitis (leukocytoclastic, isolated cerebral, systemic small vessel) Glomerulonephritis Hemolytic anemia SLE, dermatomyositis | Lymphomas Vascular malformations: peripheral, cerebral, or coronary | Elevated IgE Eosinophilia RAST: normal or positive to food antigens DPT: perivascular dermatitis with eosinophilia, spongiosis | STAT3 gene mutations (70%) Autosomal dominant Unknown (30%). |
| Autosomal recessive hyper-IgE | Moderate-to-severe eczema Urticaria Pyodermatitis Abscesses Mucocutaneous candidiasis Viral infections (HSV, HPV, molluscum, etc) | Asthma (30%-50%), rhinitis Pneumonia uncommon | Postnatal growth retardation | Vasculitis (leukocytoclastic, isolated cerebral, systemic small vessel) Glomerulonephritis Hemolytic anemia | Cancer: 17% (hematological, epithelial, others) Central nervous system: SAH, encephalopathy, stroke, facial paralysis | Elevated IgE Extreme eosinophilia Progressive lymphopenia RAST: positive to food allergens DPT: perivascular dermatitis with abundant eosinophils, spongiosis | DOCK8 gene mutations Others (rare): TYK2, PGM3 gene mutations, unknown Autosomal recessive |
| Wiskott-Aldrich syndrome | Moderate-to-severe eczema Seborrheic dermatitis Piodermitis Viral infection Petechaie, purpura | Recurrent pneumonia | Hemolytic anemia Vasculitis Nephritis Arthritis IBD | Bleeding Lymphomas: 13% | Elevated IgE and IgA Thrombocytopenia normal/low BC and TC RAST: normal | WAS gene mutations X-linked | |
| Omenn syndrome | Seborrheic dermatitis Erythroderma Eczemas (uncommon) Diffuse alopecia | Recurrent pneumonia | Postnatal growth retardation | Hepatosplenomegaly Adenopathies | Chronic multifactorial diarrhea syndrome | Elevated IgE Eosinophilia B cells absent Anomalous TC clone RAST: normal | RAG1-2 gene mutations. Less often: RMRP, ADA, IL2RG, IL7RA, DCLRE1C, CHD7, LIG4, AK2 Autosomal recessive |
| Netherton syndrome | Moderate-to-severe eczema Urticaria Pyodermatitis Ichthyosiform erythroderma; ichthyosis linearis circumflexa Trichorrhexis invaginata | Allergic asthma, rhinitis | Postnatal growth retardation Short stature | Chronic multifactorial diarrhea syndrome | Elevated IgE TC differentiation abnormality RAST: positive to food allergens DPT: psoriasiform dermatitis, hypogranulosis/agranulosis, parakeratosis, premature lamellar body secretion, subcorneal acantholysis IHC: LEKTI absence in granular layer and inner root sheaf | SPINK5 gene mutations Autosomal recessive | |
| Peeling skin syndrome, type B | Ichthyosiform erythroderma Urticaria, angioedema Pyodermatitis (Staphylococcus aureus) | Allergic asthma, rhinitis | Postnatal growth retardation | Elevated IgE Eosinophilia RAST: positive to food allergens DPT: perivascular dermatitis, subcorneal acantholysis IHC: absence CDSN in epidermis | CDSN gene mutations Autosomal recessive | ||
| Severe dermatitis, multiple allergies, metabolic wasting syndrome | Congenital erythroderma, peeling Palmoplantar keratoderma striata Hypotrichosis | Pneumonia Upper respiratory tract infections | Postnatal growth retardation | Eosinophilic esophagitis | Metabolic wasting Gastroesophageal reflux Minor cardiac defects | Elevated IgE RAST: positive to food allergens DPT: subcorneal and intragranular acantholysis, psoriasiform dermatitis, orthokeratosis/parakeratosis, hypogranulosis/hypergranulosis DIF: anomalous DSG1 cytoplasmic distribution | DSG1, DSP gene mutations Autosomal recessive |
| Prolidase deficiency | Skin fragility, ulcers (legs, perianal) Purpura, telangiectasias Photosensitivity Acute eczema Typical facies Poliosis | Recurrent upper airway infection Pneumonia | Joint hypermobility Short stature Osteoporosis | SLE | Splenomegalia Mental retardation Less often: deafness, hepatomegalia | Elevated IgE/hypergammaglobulinemia Microcytic anemia Thrombocytopenia Imidodipeptiduria | PEPD gene mutations Autosomal recessive (very variable age and phenotype) |
| Loeys-Dietz syndrome | Marfanoid appearance, cleft palate Fine skin, anomalous healing Mild eczema/occasionally severe | Pneumonia Rhinitis | Hypermobility I: bone abnormalities (cranium, sternum, phalanges, etc.) III: Osteoarthritis | Arterial aneurysms Aortic dissection | Normal/elevated IgE RAST: normal/positive to food and respiratory allergens | TGFBR1, TGFBR2, SMAD3 gene mutations Autosomal dominant | |
| Pentasomy X | Typical facies Mild eczema/occasionally severe | Recurrent pneumonia | Cranial malformations (microcephaly) Syndactylia, other bone malformations | Cardiovascular malformations Gonadal dysfunction Mental retardation | Normal/elevated IgE | Pentasomy X (no maternal disjunction in meiosis) | |
| IPEX syndrome | Severe atopic dermatitis Uncommon: erythroderma, exfoliative dermatitis, psoriasiform dermatitis, pemphigoid nodularis | Pneumonia uncommon | Enteropathy Endocrine disorders: DM-1, thyroiditis, others Hemolytic anemia Thrombocytopenia Neutropenia Hepatitis Tubular nephropathy | Sometimes severe systemic infections due to immunosuppression | Elevated IgE Elevated/normal IgA Eosinophilia Cytopenias Autoantibodies (pancreatic, thyroid, intestinal mucosa antigens, etc) | FOXP3 gene mutations X-linked | |
| STAT5B deficiency | Severe eczema Pyodermatitis Severe viral infection (VZV) Craniofacial dysmorphia Thin, fine hair | Infectious pneumonia Interstitial lymphocytic pneumonia | Dwarfism | Thyroid disease Enteropathy Interstitial lymphocytic pneumonia | GH insensitivity syndrome | Hypergammaglobulinemia Lymphopenia (TC) Normal/elevated GH Decreased IGF1, IGFBP3 Autoantibodies (thyroid, intestinal mucosa antigens, others) | STAT5B gene mutations Autosomal recessive |
BC, B cells; DIF, direct immunofluorescence; DM, diabetes mellitus; DPT, dermatopathology; DSG1, desmoglein 1; GH, growth hormone; HPV, human papillomavirus; HSV, herpes simplex virus; IBD, inflammatory bowel disease; IGF-1, insulin growth factor, type 1; IGFBP3, insulin growth factor binding protein 3; IHC, immunohistochemistry; IPEX, immune-polyendocrinopathy-enteropathy, X-linked dysregulation syndrome; RAST, radioallergoasorbent test; SAH, subarachnoid hemorrhage; SLE, systemic lupus erythematosus; TC, T cells; VZV, varicella-zoster virus.
It should be noted from the outset that the skin manifestations have a very early onset and many of these are common to several entities. Dermatitis (erythema, intense pruritus, dry skin, and lichenification) is moderate or severe in both types of hyper-IgE syndromes,2–5 Wiskott-Aldrich syndrome,6,7 Netherton syndrome,6,8 SAM,6 IPEX syndrome,3,9 and STAT5 deficiency,9,10 but is less frequent in PSS-B,6 Loeys-Dietz syndrome,11 and prolidase deficiency.12 Generally, these conditions follow a severe course with poor response to the traditional topical and systemic treatments used in atopic dermatitis. However, seborrheic dermatitis-like lesions are much more common in Omenn syndrome. In all entities, erythroderma may present with variable frequency, and even in Netherton syndrome and PSS-B, ichthyosiform erythroderma will be a dominant feature of the clinical picture and will present at a very early stage.13 The differential diagnosis should include all these possible causes of erythroderma in children. It should also be mentioned that cases of IPEX syndrome with psoriasiform dermatitis, exfoliative dermatitis, and pemphigoid nodularis have been reported.9
From the dermatopathological point of view, we can find features of great help in the diagnosis of these diseases. Perivascular dermatitis with abundant eosinophils and spongiosis is common in hyper-IgE syndromes. Histological study of skin biopsy in Netherton syndrome is characterized by hypogranulosis, parakeratosis, and subcorneal detachment, while premature secretion of lamellar bodies is also observed in electron microscopy; immunohistochemical staining demonstrates the absence of LEKTI in the granular layer and inner root sheaf. This is all a result of abnormal differentiation and accelerated scaling of the epidermis in this disease.6,8 In contrast, skin histology in PSS-B is noteworthy because of the presence of subcorneal acantholysis, superficial inflammatory infiltrate, and lack of expression of epidermal CDSN in the immunohistochemical study. Electron microscopy demonstrates loss of corneodesmosomes.6 Finally, in SAM, subcorneal and intragranular acantholysis and psoriasiform dermatitis alternating with orthoparakeratosis/parakeratosis and hypogranulosis or hypergranulosis are also found.6,14 Electron microscope study shows the irregular distribution of desmosomes in the upper half of the epidermis, with normal follicular histology.6,14 Direct immunofluorescence shows the anomalous cytoplasmic distribution of desmoglein 1 in epidermal keratinocytes.14
A predisposition to skin infections is present in all entities, although with certain particularities. The most frequent are pyodermatitis and cutaneous abscesses, as reflected in Table 1. Mucocutaneous candidiasis appears in hyper-IgE syndromes.15 In AR hyper-IgE syndrome, as well as in Wiskott-Aldrich syndrome, viral infections such as molluscum contagiosum and HPV, are also common.2–4 STAT5B deficiency has frequently been associated with severe cases of chicken pox or herpes zoster.10
Apart from these common characteristics, there are specific cutaneomucosal and morphological manifestations of certain entities which, if present, can greatly facilitate clinical diagnosis. Typical facies is often present with AD hyper-IgE syndrome5 (but not in the AR variant), Loeys-Dietz syndrome,7 Omenn syndrome, prolidase deficiency, and STAT5B deficiency.9,10 Purpuric lesions are characteristic of Wiskott-Aldrich syndrome and, less frequently, prolidase deficiency, generally in form of petechiae and ecchymosis after minimal trauma. In SAM palmoplantar keratoderma striata, skin erosions, substantial peeling, and hypotrichosis are often present.6
The skin fragility characteristic of prolidase deficiency is responsible for the development, in addition, of ulcers, predominantly on the lower limbs and, less frequently, in the anogenital region. These are generally recurrent, with an onset in early infancy, and with partial response to suitable topical treatment.12
Abnormalities in the hair follicles are varied. In Netherton syndrome, trichorrhexis invaginata (bamboo hair) is observed. This is more apparent after the first year of life and is a key characteristic to differentiate this entity from PSS-B, which is phenotypically very similar although children have normal hair.6,8 STAT5B deficiency is associated with sparse fine hair,9 and some cases of alopecia areata universalis have been reported in IPEX syndrome.10 Diffuse alopecia may develop in Omenn syndrome and very early greying may present in patients with prolidase deficiency, with diffuse alopecia in adults.12
Second, respiratory manifestations are frequent in all these children. In prolidase deficiency, of note is the high frequency of upper respiratory infections, which may help differential diagnosis with hyper-IgE syndromes.12 In fact, in AD hyper-IgE syndrome, the development of pneumatocele as a result of recurrent lung infections is relatively common.5 Recurrent pneumonia typically appears in all entities, except in Netherton syndrome and PSS-B, in which allergic asthma is more characteristic.16 In IPEX syndrome, infections are uncommon3 and are often a side effect of immunosuppressive treatment. Finally, rhinitis occurs principally in Loeys-Dietz syndrome.11
Third, we have musculoskeletal manifestations, which are key for differential diagnosis. Thus, AD hyper-IgE syndrome is associated with osteopenia, scoliosis, and pathological fractures in more than half of those affected3; however, in the AR form, postnatal growth retardation is the only musculoskeletal manifestation in some cases. On the other hand, PSS-B6 and SAM6 also show a delayed growth with short final stature, whereas in Omenn syndrome,17 prolidase deficiency,12 and Netherton syndrome,8 the delayed growth is limited, uncommon, and children will not necessarily have a short adult stature. Prolidase deficiency can be associated at times with osteoporosis and joint hypermobility.12 Loeys-Dietz syndrome11 usually presents with marfanoid features and joint hypermobility, and in type i, bone abnormalities may also present, including craniosynostosis, pectus excavatum, pectus carinatum, cavus foot, and joint contractures. In Loeys-Dietz syndrome type ii, bone abnormalities are mild or absent. The type iii syndrome is essentially associated with multifocal osteoarthritis. Pentasomy X is associated with cranial malformations, often microcephaly, as well as other bone abnormalities such as syndactylia.18 Finally, dwarfism may occur with STAT5B deficiency from birth as a result of growth hormone (GH) receptor failure, which is known as GH insensitivity syndrome.9
We will now review the main laboratory findings to discriminate between these diseases. Determination of immunoglobulins is key. Elevated IgE is observed in all entities, although we should remember that IgE levels may be normal in some patients with Loeys-Dietz syndrome and pentasomy X.11,19 In Wiskott-Aldrich syndrome and IPEX syndrome, IgA is also elevated, and in prolidase deficiency and STAT5B deficiency, polyclonal hypergammoglobulinemia is common.11,19 Multiple autoantibodies such as antiperoxidase and antivillin antibodies, among others, will be observed in IPEX syndrome and STAT5B deficiency.10
The hematology parameters may also show notable abnormalities. Eosinophilia would point towards hyper-IgE syndromes,2–5 Omenn syndrome,17 PSS-B,6 and IPEX syndrome.9 Eosinophilia is usually more marked in the recessive variant of hyper-IgE syndrome, and progressive lymphopenia is also observed. The absence of B cells is a key finding for the diagnosis of Omenn syndrome. In Wiskott-Aldrich syndrome,7 prolidase deficiency,12 and IPEX syndrome,9 thrombocytopenia, and occasionally lymphopenia are observed. With regards red blood cell parameters, hemolytic anemia is present in IPEX syndrome,9 Wiskott-Aldrich syndrome,7 and, to a lesser extent, hyper-IgE syndromes.3,5 Hypochromic microcytic anemia is most characteristic of prolidase deficiency, and here we should also mention massive iminodipeptiduria along with elevated iminodipeptides (with proline or hydroxyproline at the carboxy-terminal end) as key laboratory findings pointing to diagnosis.12 In turn, and very characteristically, in STAT5B deficiency, we can find T cell lymphopenia along with low levels of IGF1 and IGFBP3 and normal or elevated GH.10
Manifestations of atopy and allergy are characteristic of AR hyper-IgE syndrome,20 Netherton syndrome, PSS-B, SAM, and often Loeys-Dietz syndrome.15,19 The most frequent such manifestations are rhinitis and episodes of urticaria. In PSS-B, and above all in hyper-IgE syndrome, episodes of anaphylaxis and angioedema have been reported. These may be recurrent. In SAM, food allergies are usually particularly severe.16 The radioallergosorbent test, an analytical technique that quantitatively determines specific IgE levels for a range of allergens, is usually abnormal in all these entities, particularly in the case of alimentary antigens, and more frequently in individuals with atopic dermatitis.16,19 Pathological radioallergosorbent test results are less common in AD hyper-IgE syndrome than in the recessive form.19,20
Finally, we highlight other manifestations that do not fit into the above categories and that might be useful in the differential diagnosis.
The clinical picture in IPEX syndrome and STAT5B deficiency is dominated by autoimmune phenomena.9,10 In the case of IPEX syndrome, type 1 diabetes mellitus, thyroid disease, and autoimmune enteropathy are almost always present. In addition, hemolytic anemia, thrombocytopenia, leukopenia, hepatitis, and tubular nephropathy may also be present. In STAT5B deficiency, enteropathy, thyroid disease, and lymphocytic interstitial pneumonitis are the most characteristic. Autoimmune manifestations also occur in Wiskott-Aldrich syndrome (in up to 40% of patients),6,7 and hyper-IgE syndromes,3,5,20 above all in AR forms; they are less common in AD forms.20 Hemolytic anemia, leukocytoclastic vasculitis, isolated cerebral vasculitis, systemic small-vessel vasculitis, arthritis, and autoantibody-mediated glomerulonephritis can be present in hyper-IgE syndromes and Wiskott-Aldrich syndrome and, in the case of Wiskott-Aldrich syndrome, inflammatory bowel disease may also occur.6,7 In hyper-IgE syndromes, isolated cases associated with systemic lupus erythematosus and dermatomyositis have also been reported.20 In addition, an increased risk of cancer at any age has been documented for these 3 entities. Specifically, in AD hyper-IgE syndrome3,20 and Wiskott-Aldrich syndrome,6,7 lymphoproliferative diseases predominate. In AR hyper-IgE syndrome,20 a higher risk ratio has been reported with respect to the AD variant, and this higher risk is also present for neoplasms of epithelial and other origins.4
The appearance of neurological manifestation, such as hemiplegia, encephalopathy, strokes, and facial paralysis may point to AR hyper-IgE syndrome.4 However, the delay in psychomotor development, which may be very variable, is mainly associated with pentasomy X18 and prolidase deficiency.12
A manifestation common to IPEX syndrome, STAT5B deficiency, Netherton syndrome, and Omenn syndrome is chronic diarrhea syndrome.3,9,10,17 In the latter 2 entities, hepatosplenomegaly and inflammatory adenopathies are also often reported. The esophagus is affected more often in SAM6 in the form of eosinophilic esophagitis and gastroesophageal reflux.
From the cardiovascular point of view, it is important to note that arterial aneurysms and episodes of aortic dissection are characteristic of Loeys-Dietz syndrome.11 In more than 80% of cases of AD hyper-IgE syndrome, peripheral, cerebral, and, to a lesser extent, coronary vascular malformations are present.4,5 SAM is sometimes associated with minor cardiac defects,6 whereas cardiovascular abnormalities can be very varied in the case of pentasomy X.17
With regards genital abnormalities, gonadal dysfunction is only characteristic of pentasomy X18 although the external genitals are normal.
Figure 1 shows a diagnostic algorithm based on laboratory results (presence of eosinophilia and abnormalities in the radioallergosorbent test) and preliminary classification of the patients to subsequently study the presence of other findings that may guide clinical diagnosis for each of the genetic disorders discussed.

In conclusion, this article aims to provide practical assistance in the differential diagnosis of a child with refractory eczema and elevated IgE. The medical history, physical exploration, and laboratory results can guide us to the most appropriate genetic study for a definitive molecular diagnosis. This will enable individualized treatment according to the most recent evidence available for each of these entities.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
The authors would like to thank Dr. Azaña Defez for his support and guidance with the present study.
Please cite this article as: Arjona Aguilera C, Albarrán Planelles C, Tercedor Sánchez J. Trastornos genéticos con eccema moderado-grave refractario y elevación de inmunoglobulina E: diagnóstico diferencial. Actas Dermosifiliogr. 2016;107:116–124.



