


La asociación de eccema moderado-grave y niveles elevados de IgE en plasma es característica no solo de la dermatitis atópica, sino también de diversas genodermatosis: síndromes hiper-IgE, síndrome de Omenn, síndrome de Netherton, síndrome de la piel exfoliada tipo B, síndrome de dermatitis grave-alergias múltiples-desgaste metabólico, síndrome de Wiskott-Aldrich, déficit de prolidasa, síndrome de Loeys-Dietz, síndrome IPEX, déficit de STAT5B y pentasomía X. Se trata de pacientes pediátricos que presentan un cuadro clínico compatible con dermatitis atópica grave, con mala respuesta a los tratamientos clásicos y que asocian elevación de IgE desde el nacimiento. Además, comparten con frecuencia otras manifestaciones clínicas y analíticas, lo cual dificulta el diagnóstico. Presentamos una guía práctica para orientar el diagnóstico diferencial entre todas estas entidades y, por lo tanto, ayudar a decidir cuándo y el tipo de test genético a realizar para establecer el diagnóstico definitivo.
The association of moderate to severe eczema and elevated plasma levels of immunoglobulin E is a characteristic not only of atopic dermatitis but also of various genodermatoses: hyperimmunoglobulin E syndromes, Omenn syndrome, Netherton syndrome, peeling skin syndrome type B, severe dermatitis, multiple allergies, and metabolic wasting syndrome, Wiskott-Aldrich syndrome, prolidase deficiency, Loeys-Dietz syndrome, IPEX syndrome, STAT5B deficiency, and pentasomy X. The clinical presentation of these genodermatoses –typically in children– is consistent with severe atopic dermatitis. Immunoglobulin E is elevated from birth and response to conventional treatments is poor. Diagnosis is further complicated by the fact that these genodermatoses often share other clinical manifestations and laboratory findings. We present practical guidelines for differentiating among these various entities, with the aim of helping physicians decide what type of genetic test should be carried out –and when– in order to establish a definitive diagnosis.
La asociación de eccema moderado-grave y niveles elevados de IgE en plasma es característica de la dermatitis atópica, pero también se manifiesta típicamente en varias enfermedades genéticas. Muchas de ellas asocian una inmunodeficiencia congénita concomitante. Ante un enfermo con clínica compatible con dermatitis atópica grave1 que no responde al tratamiento clásico de esta (medidas higiénico-dietéticas, corticoides tópicos o sistémicos, ciclosporina, metotrexato u azatioprina)1, junto con elevación de inmunoglobulina E (IgE) desde sus primeros días de vida, podemos sospechar la existencia de uno de estos síndromes.
Por una parte, se han descrito los síndromes hiper-IgE, extensamente estudiados a día de hoy y que se clasifican en función de su patrón de herencia: autosómica dominante (AD) y autosómica recesiva (AR). Ambas se caracterizan por una IgE elevada y un cuadro clínico dominado por la afectación cutánea, con eccemas agudos y subagudos moderados o graves que recuerdan a la dermatitis atópica, y por las infecciones cutáneas y respiratorias recurrentes.
Por otro lado, existe una serie de enfermedades congénitas de baja prevalencia que presentan con mucha frecuencia episodios de eccemas rebeldes asociados a la elevación de IgE y debemos incluir en nuestro algoritmo diagnóstico. El síndrome de Omenn es una inmunodeficiencia combinada grave de herencia AR que se manifiesta antes del año de vida. El síndrome de Wiskott-Aldrich constituye una inmunodeficiencia primaria de herencia ligada al X caracterizado fundamentalmente por diátesis hemorrágica secundaria a trombocitopenia o disfunción plaquetaria, junto con eccemas e infecciones bacterianas recurrentes desde el nacimiento. El déficit de prolidasa es una enfermedad genética AR multisistémica con una clínica y gravedad muy variables, que asocia actividad escasa o ausente de la enzima prolidasa en eritrocitos, leucocitos o fibroblastos. El síndrome de Loeys-Dietz se trata de una conectivopatía hereditaria AD que asocia un aspecto marfanoide y anomalías en grandes arterias. El síndrome de disregulación inmune-poliendocrinopatía-enteropatía ligada a X, más conocido como síndrome IPEX (S-IPEX), es una enfermedad autoinmune congénita grave en la cual, además de la afectación cutánea, destacan diarrea refractaria, infecciones y endocrinopatías múltiples. El déficit de STAT5B (DSTAT5B) se incluye en el grupo de síndromes de insensibilidad a la hormona de crecimiento, que destaca por asociar una inmunodeficiencia primaria grave de herencia AR. La pentasomía X es una enfermedad congénita por alteración cromosómica en mujeres que origina disfunción gonadal, retraso del desarrollo, estatura baja y anomalías musculoesqueléticas y craneofaciales. El síndrome de Netherton constituye una genodermatosis AR caracterizada por eritrodermia ictiosiforme, tricorrexis invaginada y manifestaciones atópicas desde prácticamente el nacimiento. Finalmente, incluimos 2 genodermatosis relacionadas con el síndrome de Netherton desde el punto de vista fisiopatológico de muy reciente descripción: el síndrome de la piel exfoliada tipo B (PSS-B) y el síndrome de dermatitis grave-alergias múltiples-desgaste metabólico (SAM). El PSS-B es una genodermatosis AR con eritrodermia ictiosiforme desde el nacimiento que, además, presenta alergias alimentarias graves, angioedema y urticaria. Finalmente, el SAM es otra genodermatosis AR rara donde la afectación cutánea se combina con alergias alimentarias importantes, afectación esofágica y datos característicos que se comentan a lo largo del artículo.
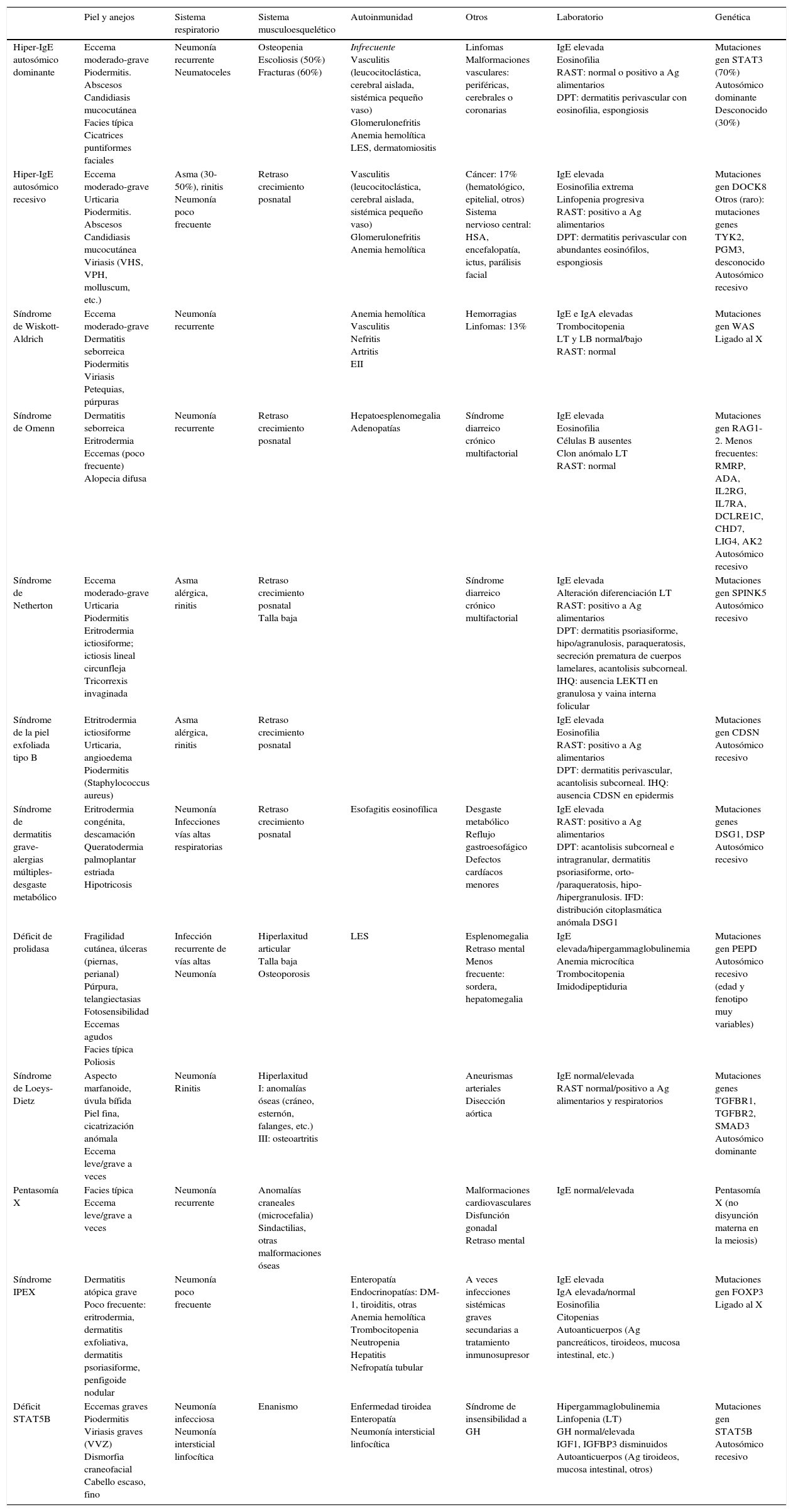
El diagnóstico diferencial entre todos estos síndromes puede resultar muy complejo, dado que pueden solaparse a menudo, compartiendo muchas de sus características clínicas. La expresión fenotípica de cada una de estas entidades puede ser variable en cada individuo. Por todo ello, en el presente trabajo aportamos aquellos datos clave que permitan realizar el diagnóstico diferencial desde el punto de vista clínico, a partir del cual podamos orientar el estudio genético a solicitar. El diagnóstico definitivo lo otorga la identificación del gen causal mutado. En la tabla 1 hemos recogido las principales genodermatosis que debemos incluir en el diagnóstico diferencial, señalando las principales características a investigar en el proceso diagnóstico que nos van a permitir individualizarla. La descripción completamente detallada de cada una de estas enfermedades no es el objetivo de este artículo, y se remite al lector a utilizar la bibliografía para obtener mayor información.
Diagnóstico diferencial clínico y analítico entre genodermatosis que manifiestan eccema moderado-grave y elevación de IgE
| Piel y anejos | Sistema respiratorio | Sistema musculoesquelético | Autoinmunidad | Otros | Laboratorio | Genética | |
|---|---|---|---|---|---|---|---|
| Hiper-IgE autosómico dominante | Eccema moderado-grave Piodermitis. Abscesos Candidiasis mucocutánea Facies típica Cicatrices puntiformes faciales | Neumonía recurrente Neumatoceles | Osteopenia Escoliosis (50%) Fracturas (60%) | Infrecuente Vasculitis (leucocitoclástica, cerebral aislada, sistémica pequeño vaso) Glomerulonefritis Anemia hemolítica LES, dermatomiositis | Linfomas Malformaciones vasculares: periféricas, cerebrales o coronarias | IgE elevada Eosinofilia RAST: normal o positivo a Ag alimentarios DPT: dermatitis perivascular con eosinofilia, espongiosis | Mutaciones gen STAT3 (70%) Autosómico dominante Desconocido (30%) |
| Hiper-IgE autosómico recesivo | Eccema moderado-grave Urticaria Piodermitis. Abscesos Candidiasis mucocutánea Viriasis (VHS, VPH, molluscum, etc.) | Asma (30-50%), rinitis Neumonía poco frecuente | Retraso crecimiento posnatal | Vasculitis (leucocitoclástica, cerebral aislada, sistémica pequeño vaso) Glomerulonefritis Anemia hemolítica | Cáncer: 17% (hematológico, epitelial, otros) Sistema nervioso central: HSA, encefalopatía, ictus, parálisis facial | IgE elevada Eosinofilia extrema Linfopenia progresiva RAST: positivo a Ag alimentarios DPT: dermatitis perivascular con abundantes eosinófilos, espongiosis | Mutaciones gen DOCK8 Otros (raro): mutaciones genes TYK2, PGM3, desconocido Autosómico recesivo |
| Síndrome de Wiskott-Aldrich | Eccema moderado-grave Dermatitis seborreica Piodermitis Viriasis Petequias, púrpuras | Neumonía recurrente | Anemia hemolítica Vasculitis Nefritis Artritis EII | Hemorragias Linfomas: 13% | IgE e IgA elevadas Trombocitopenia LT y LB normal/bajo RAST: normal | Mutaciones gen WAS Ligado al X | |
| Síndrome de Omenn | Dermatitis seborreica Eritrodermia Eccemas (poco frecuente) Alopecia difusa | Neumonía recurrente | Retraso crecimiento posnatal | Hepatoesplenomegalia Adenopatías | Síndrome diarreico crónico multifactorial | IgE elevada Eosinofilia Células B ausentes Clon anómalo LT RAST: normal | Mutaciones gen RAG1-2. Menos frecuentes: RMRP, ADA, IL2RG, IL7RA, DCLRE1C, CHD7, LIG4, AK2 Autosómico recesivo |
| Síndrome de Netherton | Eccema moderado-grave Urticaria Piodermitis Eritrodermia ictiosiforme; ictiosis lineal circunfleja Tricorrexis invaginada | Asma alérgica, rinitis | Retraso crecimiento posnatal Talla baja | Síndrome diarreico crónico multifactorial | IgE elevada Alteración diferenciación LT RAST: positivo a Ag alimentarios DPT: dermatitis psoriasiforme, hipo/agranulosis, paraqueratosis, secreción prematura de cuerpos lamelares, acantolisis subcorneal. IHQ: ausencia LEKTI en granulosa y vaina interna folicular | Mutaciones gen SPINK5 Autosómico recesivo | |
| Síndrome de la piel exfoliada tipo B | Etritrodermia ictiosiforme Urticaria, angioedema Piodermitis (Staphylococcus aureus) | Asma alérgica, rinitis | Retraso crecimiento posnatal | IgE elevada Eosinofilia RAST: positivo a Ag alimentarios DPT: dermatitis perivascular, acantolisis subcorneal. IHQ: ausencia CDSN en epidermis | Mutaciones gen CDSN Autosómico recesivo | ||
| Síndrome de dermatitis grave-alergias múltiples-desgaste metabólico | Eritrodermia congénita, descamación Queratodermia palmoplantar estriada Hipotricosis | Neumonía Infecciones vías altas respiratorias | Retraso crecimiento posnatal | Esofagitis eosinofílica | Desgaste metabólico Reflujo gastroesofágico Defectos cardíacos menores | IgE elevada RAST: positivo a Ag alimentarios DPT: acantolisis subcorneal e intragranular, dermatitis psoriasiforme, orto-/paraqueratosis, hipo-/hipergranulosis. IFD: distribución citoplasmática anómala DSG1 | Mutaciones genes DSG1, DSP Autosómico recesivo |
| Déficit de prolidasa | Fragilidad cutánea, úlceras (piernas, perianal) Púrpura, telangiectasias Fotosensibilidad Eccemas agudos Facies típica Poliosis | Infección recurrente de vías altas Neumonía | Hiperlaxitud articular Talla baja Osteoporosis | LES | Esplenomegalia Retraso mental Menos frecuente: sordera, hepatomegalia | IgE elevada/hipergammaglobulinemia Anemia microcítica Trombocitopenia Imidodipeptiduria | Mutaciones gen PEPD Autosómico recesivo (edad y fenotipo muy variables) |
| Síndrome de Loeys-Dietz | Aspecto marfanoide, úvula bífida Piel fina, cicatrización anómala Eccema leve/grave a veces | Neumonía Rinitis | Hiperlaxitud I: anomalías óseas (cráneo, esternón, falanges, etc.) III: osteoartritis | Aneurismas arteriales Disección aórtica | IgE normal/elevada RAST normal/positivo a Ag alimentarios y respiratorios | Mutaciones genes TGFBR1, TGFBR2, SMAD3 Autosómico dominante | |
| Pentasomía X | Facies típica Eccema leve/grave a veces | Neumonía recurrente | Anomalías craneales (microcefalia) Sindactilias, otras malformaciones óseas | Malformaciones cardiovasculares Disfunción gonadal Retraso mental | IgE normal/elevada | Pentasomía X (no disyunción materna en la meiosis) | |
| Síndrome IPEX | Dermatitis atópica grave Poco frecuente: eritrodermia, dermatitis exfoliativa, dermatitis psoriasiforme, penfigoide nodular | Neumonía poco frecuente | Enteropatía Endocrinopatías: DM-1, tiroiditis, otras Anemia hemolítica Trombocitopenia Neutropenia Hepatitis Nefropatía tubular | A veces infecciones sistémicas graves secundarias a tratamiento inmunosupresor | IgE elevada IgA elevada/normal Eosinofilia Citopenias Autoanticuerpos (Ag pancreáticos, tiroideos, mucosa intestinal, etc.) | Mutaciones gen FOXP3 Ligado al X | |
| Déficit STAT5B | Eccemas graves Piodermitis Viriasis graves (VVZ) Dismorfia craneofacial Cabello escaso, fino | Neumonía infecciosa Neumonía intersticial linfocítica | Enanismo | Enfermedad tiroidea Enteropatía Neumonía intersticial linfocítica | Síndrome de insensibilidad a GH | Hipergammaglobulinemia Linfopenia (LT) GH normal/elevada IGF1, IGFBP3 disminuidos Autoanticuerpos (Ag tiroideos, mucosa intestinal, otros) | Mutaciones gen STAT5B Autosómico recesivo |
Ag: antígeno; DM-1: diabetes mellitus tipo 1; DPT: dermatopatología; DSG1: desmogleína 1; EII: enfermedad inflamatoria intestinal; GH: hormona del crecimiento; HSA: hemorragia subaracnoidea; IFD: inmunofluorescencia directa; IGF-1: factor de crecimiento insulínico tipo 1; IGFBP3: proteína 3 de unión al factor de crecimiento insulínico; IHQ: inmunohistoquímica; IPEX: síndrome de disregulación inmune-poliendocrinopatía-enteropatía ligada a X; LB: linfocitos B; LES: lupus eritematoso sistémico; LT: linfocitos T; RAST: radioallergosorbent test; VHS: virus del herpes simple; VPH: virus del papiloma humano; VVZ: virus varicela zóster.
En primer lugar, las manifestaciones cutáneas son de aparición muy precoz, y muchas de ellas son comunes a varias entidades. La dermatitis (eritema, prurito intenso, sequedad cutánea y liquenificación) se manifiesta de forma moderada o grave en ambos tipos de síndromes hiper-IgE2–5, síndrome de Wiskott-Aldrich6,7, síndrome de Netherton6,8, SAM6, S-IPEX3,9 y DSTAT59,10, siendo menos frecuente en el PSS-B6, síndrome de Loeys-Dietz11 y déficit de prolidasa12. Generalmente su curso es grave, con mala respuesta a los tratamientos tópicos y sistémicos clásicos de la dermatitis atópica. Sin embargo, en el síndrome de Omenn las lesiones tipo dermatitis seborreica son mucho más frecuentes. En todas las entidades puede llegar a producirse eritrodermia con frecuencia variable, e incluso en el síndrome de Netherton y PSS-B la eritrodermia ictiosiforme va a ser el cuadro clínico cutáneo predominante y de aparición muy precoz13. El diagnóstico diferencial deberá incluir todas aquellas posibles causas de eritrodermia en niños. Además, cabe señalar la descripción en el S-IPEX de algunos casos con dermatitis psoriasiforme, dermatitis exfoliativa o penfigoide nodular9.
Desde el punto de vista dermatopatológico, podemos encontrar datos diferenciales de gran ayuda al diagnóstico en varias de estas enfermedades. Así, en los síndromes hiper-IgE se encuentra frecuentemente una dermatitis perivascular con abundantes eosinófilos y espongiosis. El estudio histológico de la biopsia cutánea en el síndrome de Netherton se caracteriza por hipogranulosis, paraqueratosis y despegamiento subcórneo, observándose además secreción prematura de cuerpos lamelares con microscopia electrónica; la inmunohistoquímica demuestra ausencia de LEKTI en la capa granulosa y la vaina interna folicular. Todo ello es consecuencia de la diferenciación anormal y la descamación acelerada de la epidermis que ocurre en esta enfermedad6,8. Por otra parte, la histología cutánea en el PSS-B destaca por presentar acantolisis subcorneal, infiltrado inflamatorio superficial y ausencia de expresión de CDSN epidérmica en el estudio inmunohistoquímico. La microscopia electrónica demuestra la pérdida de corneodesmosomas6. Finalmente, en el SAM suele encontrarse acantolisis subcorneal e intragranular y dermatitis psoriasiforme alternando orto/paraqueratosis e hipo/hipergranulosis6,14. El estudio por microscopia electrónica pone de manifiesto la distribución irregular de desmosomas en la mitad superior de la epidermis, siendo la histología folicular normal6,14. La inmunofluorescencia directa demuestra la distribución citoplasmática anómala de desmogleína 1 en los queratinocitos epidérmicos14.
La predisposición a infecciones cutáneas se manifiesta en todos los cuadros, aunque con algunas particularidades. Lo más frecuente son las piodermitis y los abscesos cutáneos, como vemos en la tabla 1. La candidiasis mucocutánea aparece en los síndromes hiper-IgE15. En el síndrome hiper-IgE AR, así como en el síndrome de Wiskott-Aldrich, son además frecuentes las viriasis, tales como el molluscum contagiosum o el VPH2–4. Por su parte, el DSTAT5B se ha asociado con frecuencia a cuadros graves de varicela o herpes zóster10.
Aparte de estas características comunes, existen manifestaciones cutaneomucosas y morfológicas específicas de algunos cuadros que, de estar presentes, van a facilitar mucho el diagnóstico clínico. Una facies típica suele observarse en el síndrome hiper-IgE AD5 (no en su variante AR), el síndrome de Loeys-Dietz7, el síndrome de Omenn, el déficit de prolidasa y el DSTAT5B9,10. Las lesiones purpúricas son características del síndrome de Wiskott-Aldrich y, con menor frecuencia, del déficit de prolidasa, generalmente en forma de petequias y equimosis en la piel ante mínimos traumatismos. En el SAM6 aparece típicamente una queratodermia palmoplantar estriada, erosiones cutáneas, descamación importante e hipotricosis.
La fragilidad cutánea característica del déficit de prolidasa hace que los enfermos desarrollen, además, úlceras de predominio en los miembros inferiores y, con menor frecuencia, en la región anogenital, generalmente recurrentes, de inicio en la infancia temprana y con respuesta parcial a los tratamientos tópicos adecuados12.
Las alteraciones del folículo piloso son variadas. En el síndrome de Netherton se observa tricorrexis invaginada («pelos de bambú»), más evidente a partir del año de vida, característica clave para diferenciarlo del PSS-B, fenotípicamente muy similar, pero el cabello es normal en estos niños6,8. El DSTAT5B asocia pelo escaso y fino9, y en el S-IPEX se ha descrito algún caso de alopecia areata universal10. El síndrome de Omenn puede desarrollar alopecia difusa, y en el déficit de prolidasa12 aparece en ocasiones canicie muy precoz, junto con alopecia difusa en la edad adulta.
En segundo lugar, las manifestaciones respiratorias son frecuentes en todos estos niños. En el déficit de prolidasa destaca la elevada frecuencia de infecciones respiratorias de vías altas, lo que puede plantear el diagnóstico diferencial con los síndromes hiper-IgE12. De hecho, en el síndrome hiperIgE AD el desarrollo de neumatoceles a consecuencia de las infecciones pulmonares repetidas es relativamente frecuente5. La neumonía recurrente aparece típicamente en todos los grupos, salvo en el síndrome de Netherton y el PSS-B, en los que el asma alérgica es lo más característico16. En el S-IPEX las infecciones son infrecuentes3 y en muchas ocasiones se consideran secundarias a tratamientos inmunosupresores. Finalmente, la rinitis ocurre principalmente en el síndrome de Loeys-Dietz11.
En tercer lugar incluimos las manifestaciones osteomusculares, clave para el diagnóstico diferencial. Así, el síndrome hiper-IgE AD3 se asocia a osteopenia, escoliosis y fracturas patológicas en más de la mitad de los enfermos; sin embargo, en la forma AR solo ocurre retraso en el crecimiento posnatal en algunos casos. Por otra parte, el PSS-B6 y el SAM6 también manifiestan un retraso del crecimiento con talla baja final, mientras en el síndrome de Omenn17, el déficit de prolidasa12 y el síndrome de Netherton8 el retraso del crecimiento es leve, poco frecuente y no necesariamente se alcanzará una talla baja final. El déficit de prolidasa12 puede asociarse en ocasiones a osteoporosis e hiperlaxitud de las articulaciones. El síndrome de Loeys-Dietz11 suele presentar un aspecto y complexión marfanoide con hiperlaxitud articular, y en el tipo i ocurren, además, múltiples alteraciones óseas, incluyendo craneosinostosis, pectus excavatum, pectus carinatum, pies cóncavos y contracturas articulares. En el síndrome de Loeys-Dietz tipo ii las anomalías óseas son leves o ausentes. El tipo iii asocia fundamentalmente osteoartritis multifocal. La pentasomía X18, por su parte, se asocia a alteraciones craneales, frecuentemente microcefalia, así como a otras anomalías óseas como sindactilia en las extremidades. Finalmente, en el DSTAT5B9 ocurre un enanismo desde el nacimiento secundario al fallo en el receptor de GH, lo que se ha denominado «síndrome de insensibilidad a GH».
A continuación revisamos las principales alteraciones analíticas a tener en cuenta para discriminar entre estas enfermedades. La determinación de inmunoglobulinas es clave. Así, veremos una IgE elevada en todos los cuadros, si bien debemos tener en cuenta que en algunos pacientes con síndrome de Loeys-Dietz y pentasomía X la IgE puede ser normal11,19. Por otra parte, en el síndrome de Wiskott-Aldrich y el S-IPEX la IgA también se eleva, y en el déficit de prolidasa y DSTAT5B es frecuente la hipergammaglobulinemia policlonal11,19. En el S-IPEX y el DSTAT5B vamos a encontrar además múltiples autoanticuerpos, como antiperoxidasa y antivillina, entre otros10.
El hemograma también puede mostrar alteraciones significativas. La eosinofilia nos orienta hacia los síndromes hiper-IgE2–5, el síndrome de Omenn17, el PSS-B6 y el S-IPEX9. En la variante recesiva del síndrome hiper-IgE la eosinofilia suele ser más marcada y además se observa linfopenia progresiva. En el síndrome de Omenn la ausencia de linfocitos B es un dato clave en su diagnóstico. Por su parte, el síndrome de Wiskott-Aldrich7, el déficit de prolidasa12 y el S-IPEX9 muestran trombocitopenia y, en ocasiones, linfopenia. En cuanto a la serie roja, la anemia hemolítica ocurre en el S-IPEX9, el síndrome de Wiskott-Aldrich7 y, con menor frecuencia, en los síndromes hiper-IgE3,5. La anemia microcítica hipocroma es más característica del déficit de prolidasa, y cabe señalar aquí la iminodipeptiduria masiva junto con la elevación de iminodipéptidos (con prolina o hidroxiprolina en el extremo carboxiterminal) como datos analíticos clave que señalan este diagnóstico12. Por su parte, y de forma muy característica, en el DSTAT5B vamos a encontrar linfopenia de células T junto con niveles bajos de IGF1 e IGFBP3 y niveles normales o elevados de GH10.
Las manifestaciones de atopia y alergia son características del síndrome hiper-IgE AR20, el síndrome de Netherton, el PSS-B, el SAM y, con frecuencia, el síndrome de Loeys-Dietz15,19. Lo más frecuente es la rinitis y episodios de urticaria. En el PSS-B, y sobre todo en el síndrome hiper-IgE AR, se han descrito además episodios de anafilaxia y angioedema que pueden ser recurrentes. En el SAM las alergias alimentarias suelen ser especialmente graves16. El radioallergosorbent test, prueba analítica que determina de forma cuantitativa el nivel de IgE específica de multitud de alérgenos, suele encontrarse alterado en todos los cuadros señalados, sobre todo frente a antígenos alimentarios, con mayor frecuencia que en la dermatitis atópica16,19. En el síndrome hiper-IgE AD el radioallergosorbent test patológico ocurre con menor frecuencia que en la forma recesiva19,20.
En último lugar queremos destacar otras manifestaciones que no se encuadran en las categorías anteriores y pueden ser muy útiles en el diagnóstico diferencial.
El cuadro clínico presentado por el S-IPEX y el DSTAT5B se debe fundamentalmente a fenómenos de autoinmunidad9,10. En el primero, la diabetes mellitus tipo 1, la enfermedad tiroidea y la enteropatía autoinmunes son casi constantes. Además, se ha asociado a anemia hemolítica, trombocitopenia, leucopenia, hepatitis y nefropatía tubular. En el DSTAT5B lo más característico es la enteropatía, la enfermedad tiroidea y la neumonitis intersticial linfocítica. Las manifestaciones autoinmunes también ocurren en el síndrome de Wiskott-Aldrich6,7 (hasta en un 40% de los enfermos) y los síndromes hiper-IgE3,5,20, sobre todo en la forma AR, siendo infrecuentes en la forma AD20. En los 3 puede ocurrir anemia hemolítica, vasculitis leucocitoclástica, vasculitis cerebral aislada, vasculitis sistémicas de pequeño vaso, artritis, glomerulonefritis mediada por autoanticuerpos y, en el caso del síndrome de Wiskott-Aldrich, también ocurre enfermedad inflamatoria intestinal6,7. En los síndromes hiper-IgE se han descrito además algunos casos asociados a lupus eritematoso sistémico y a dermatomiositis20. Por otra parte, en estas 3 entidades se ha documentado el riesgo aumentado de cáncer a cualquier edad. Concretamente, en el síndrome hiper-IgE AD3,20 y el síndrome de Wiskott-Aldrich6,7 predominan las enfermedades linfoproliferativas. En el síndrome hiper-IgE AR20 se ha descrito una proporción de riesgo mayor respecto a su variante AD, que incluye, además, neoplasias de estirpe epitelial u otras4.
La aparición de manifestaciones neurológicas, tales como encefalopatía, hemiplejia, ictus cerebrales o parálisis facial, puede orientarnos hacia un síndrome hiper-IgE AR4. Sin embargo, el retraso del desarrollo psicomotor, en grado muy variable, se asocia principalmente a la pentasomía X18 y al déficit de prolidasa12.
Una manifestación común al S-IPEX, el DSTAT5B, el síndrome de Netherton y el síndrome de Omenn es el síndrome diarreico crónico3,9,10,17. En los 2 últimos se evidencia además hepatoesplenomegalia y adenopatías de carácter inflamatorio con cierta frecuencia. El esófago se afecta con mayor frecuencia en el SAM6 en forma de esofagitis eosinofílica y reflujo gastroesofágico.
Desde el punto de vista cardiovascular, es importante señalar que los aneurismas arteriales y los episodios de disección aórtica son característicos del síndrome de Loeys-Dietz11. El síndrome hiper-IgE AD manifiesta en más del 80% de los casos malformaciones vasculares periféricas, cerebrales o, con menor frecuencia, a nivel coronario4,5. El SAM6 a veces asocia defectos cardíacos menores, mientras que en la pentasomía X17 las alteraciones cardiovasculares pueden ser muy variadas.
En cuanto a alteraciones genitales, solo la pentasomía X18 desarrolla característicamente una disfunción gonadal, preservando la normalidad de los genitales externos.
Finalmente, ofrecemos en la figura 1 un algoritmo diagnóstico partiendo de datos analíticos (presencia de eosinofilia y alteraciones en el radioallergosorbent test) como clasificación preliminar de los pacientes para, a continuación, estudiar la presencia de otros datos guía, que nos orienta a un diagnóstico clínico de cada una de los trastornos genéticos considerados.

En conclusión, presentamos un trabajo con finalidad eminentemente práctica para el diagnóstico diferencial ante un niño con eccemas rebeldes y elevación de IgE. Los datos de la anamnesis, la exploración física y los hallazgos analíticos recogidos pueden orientarnos hacia el estudio genético más adecuado que nos permitirá el diagnóstico molecular definitivo. Gracias a ello podremos ofrecer un tratamiento individualizado según la última evidencia disponible para cada una de estas entidades.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.
Los autores agradecemos al Dr. Azaña Defez su apoyo y atención ante la consulta en relación con el presente trabajo.



