

La distrofia fascial congénita (DFC) o síndrome de la piel rígida es una enfermedad cutánea rara y que fue descrita en 1971 por Esterly y McKusick1. En este proceso se produce una fibrosis no inflamatoria del tejido celular subcutáneo y la fascia muscular ocasionando un endurecimiento de la piel y una dificultad en la movilización de las articulaciones subyacentes. Puede ser hereditaria y mostrar un grado de afectación muy variable, a veces con una mínima sintomatología, como ocurrió en el caso que se describe, cuyo diagnóstico se realizó en la edad adulta.
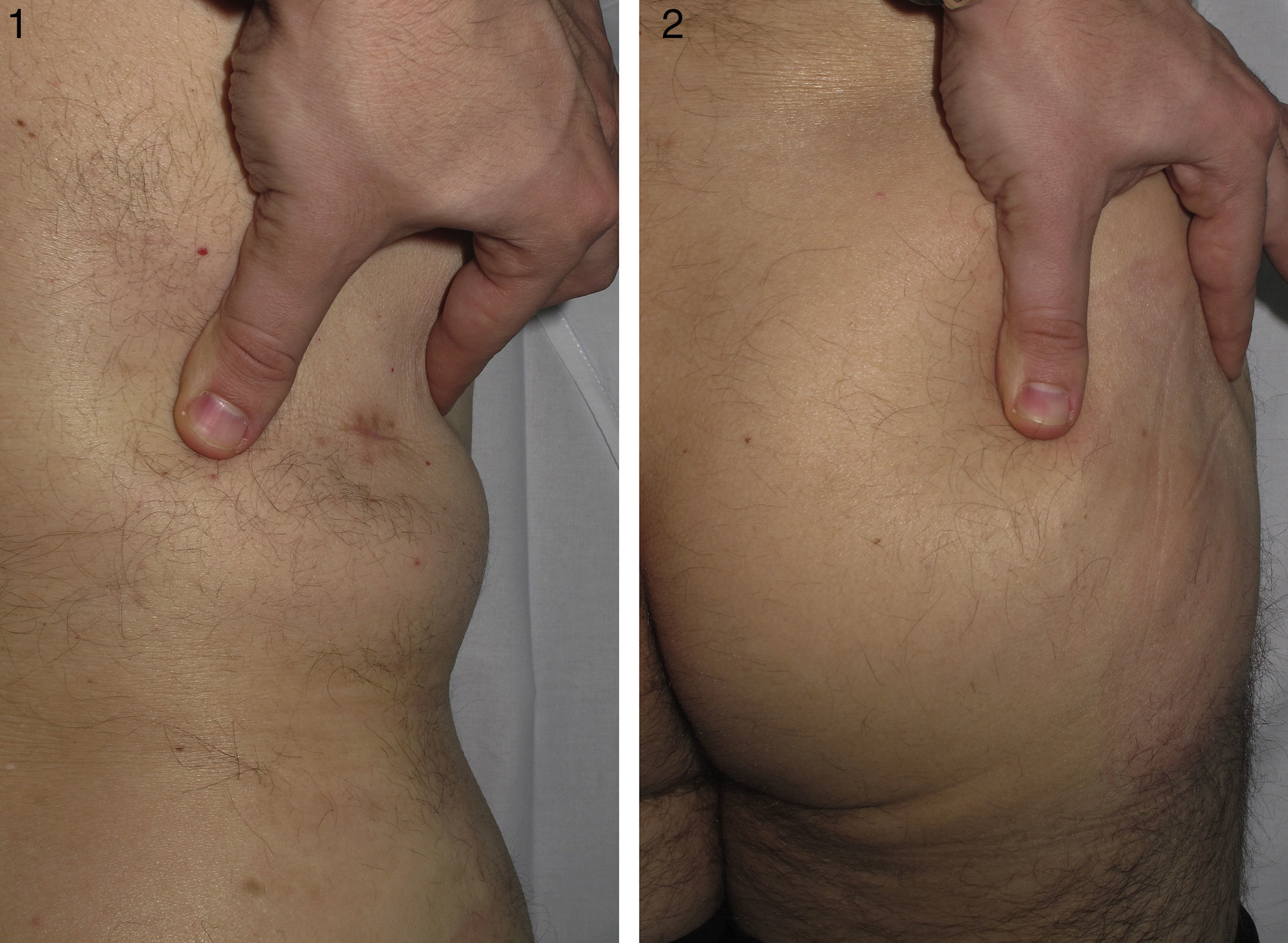
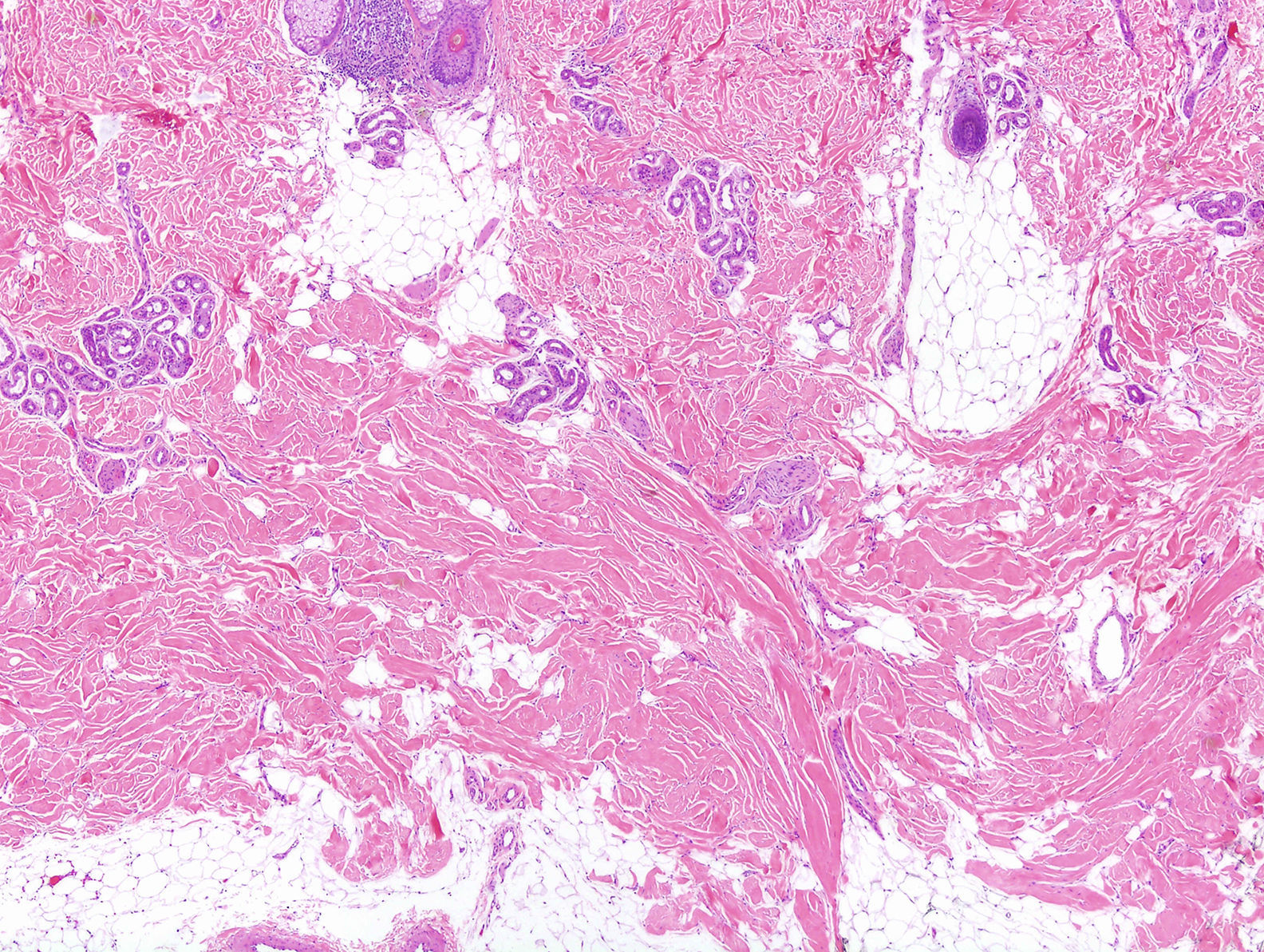
Varón de 46 años, sin antecedentes personales de interés, procedente de otro centro con sospecha clínica de morfea profunda que no había respondido al tratamiento con corticoides orales. El paciente refería, desde la infancia, dificultad en la realización de algunos movimientos, como la flexión del tronco, así como la imposibilidad de recibir inyecciones intramusculares en el glúteo derecho. También explicaba síntomas similares en su hija. En la exploración se constató la dificultad de pellizcar la piel de la zona lumbar y el glúteo derecho (figs. 1 y 2), que ofrecía un tacto duro, así como la limitación de los movimientos de la articulación coxofemoral derecha, en especial los relacionados con la flexión de la misma. En el centro anterior se habían cursado unos análisis con estudio de autoinmunidad en los que no se observaron alteraciones, una resonancia magnética en la que se descartaba que hubiera afectación ósea o muscular y una biopsia de piel informada como compatible con morfea profunda. Se revisó esta misma biopsia y en ella se observó un colágeno engrosado dispuesto de forma horizontal en las capas más profundas de la dermis, siguiendo un patrón en enrejado y una ausencia de infiltrado inflamatorio (fig. 3). Tras la correlación clínico-patológica se llegó al diagnóstico de una DFC y se aconsejó realizar fisioterapia.
. En la piel de la zona lumbar no se observaron cambios visibles, pero se pudo comprobar la dificultad de plegarla entre los dedos. 2). En la piel de la zona glútea se objetivaron cambios similares a los de la zona lumbar con una piel de tacto duro y sin cambios visibles en la superficie")
1). En la piel de la zona lumbar no se observaron cambios visibles, pero se pudo comprobar la dificultad de plegarla entre los dedos. 2). En la piel de la zona glútea se objetivaron cambios similares a los de la zona lumbar con una piel de tacto duro y sin cambios visibles en la superficie

La DFC es una entidad muy poco frecuente y escasamente descrita en la literatura2. Es un proceso que puede aparecer tanto en los hombres como en las mujeres, sin predominio por ninguno de los 2 sexos, y en el que un 30% de los pacientes tienen antecedentes familiares2. En el caso descrito el interrogatorio dirigido permitió identificar una hija con una clínica similar, si bien no se han realizado por el momento exploraciones complementarias que permitan confirmar el diagnóstico.
Hasta hace pocos años se desconocía la patogenia por la cual se desarrollaba la enfermedad3. Sin embargo, Loeys et al.4 descubrieron que todos los individuos afectos tenían una mutación en el gen de la fibrilina-1. Esta proteína, que también tienen mutada los pacientes con síndrome de Marfan, está implicada principalmente en la formación de las microfibrillas (formadas por polímeros de fibrilina) que son las que, junto con la elastina, forman las fibras elásticas. Esta mutación deriva en una acumulación desorganizada de microfibrillas en la dermis que puede observarse mediante microscopia electrónica o confocal. Estas acumulaciones producen la activación anómala de otra molécula, llamada factor de crecimiento transformante (TGF-beta), que tiene la capacidad de promover el depósito de colágeno en la dermis. En los pacientes con DFC el aumento de TGF-beta deriva en un mayor depósito de colágeno en las partes profundas de la dermis, el tejido celular subcutáneo o la fascia muscular.
El inicio de la clínica suele ser congénito, pero también puede aparecer alrededor de los primeros 6 años de vida2. Los pacientes con DFC presentan áreas de piel endurecida, con límites bien definidos y sin cambios visibles en la superficie, sobre todo en las zonas cercanas a la cintura pélvica o escapular y en la parte proximal de los muslos5. Otros hallazgos menos frecuentes son la hipertricosis, la hiperpigmentación en las áreas afectas6 o la presencia de nódulos subcutáneos en las falanges distales de los dedos de las manos4.
El principal problema que conlleva este proceso es la limitación de la movilidad articular que se deriva de él. En la mayoría de pacientes esta limitación es leve y no interfiere en exceso con la vida diaria, sin embargo, la afectación puede ser muy extensa limitando incluso la capacidad respiratoria2. Si los síntomas son leves el diagnóstico puede retrasarse hasta la vida adulta.
Además de la clínica los hallazgos microscópicos también son muy importantes. En la microscopia se objetiva una proliferación del tejido colágeno, sobre todo en la fascia muscular y el tejido celular subcutáneo, aunque también hay casos publicados con afectación exclusiva de la dermis reticular7. Es importante hacer hincapié en que la afectación de la fascia muscular no es imprescindible para el diagnóstico de esta entidad. El término «distrofia fascial congénita» se propuso en la descripción original al observar la afectación de la fascia en todos los pacientes con este diagnóstico. Sin embargo, la participación de esta estructura no se ha demostrado de forma uniforme con posterioridad. El hallazgo microscópico más característico y sugestivo de DFC no es tanto la localización del colágeno que prolifera, sino su disposición adoptando un patrón reticulado7. Otros hallazgos típicos son la ausencia de infiltrado inflamatorio y la presencia de mucina.
Finalmente, para establecer el diagnóstico es importante descartar la presencia de autoanticuerpos específicos, así como también lesión estructural ósea o muscular2.
El diagnóstico diferencial debe establecerse, principalmente, con la morfea, en su variante profunda2. A diferencia de la DFC esta suele tener una distribución asimétrica, no suele iniciarse en edades tempranas, en general muestra cambios visibles en la piel y, desde el punto de vista microscópico, el colágeno se dispone de forma compacta. Además, en las fases iniciales suele haber un infiltrado inflamatorio de tipo linfoplasmocitario. En la morfea panesclerótica de la infancia, si bien la afectación del proceso escleroso es también profunda como en la DFC, la existencia de cambios en la piel muy evidentes, en forma de ulceraciones y trastornos de la pigmentación, permite un fácil diagnóstico diferencial8.
No hay tratamiento eficaz para la DFC. Se han utilizado múltiples fármacos como corticoides tópicos y orales, metotrexato o PUVA y los resultados de todos ellos han sido muy pobres2. Solo la fisioterapia parece que podría mejorar la movilidad y comodidad en las articulaciones afectas.
El interés de conocer y diagnosticar de forma correcta la DFC radica esencialmente en ser capaces de diferenciarla de la morfea profunda, un proceso con el que guarda similitudes clínicas y microscópicas. Solo de esta forma será posible evitar la administración de fármacos con potenciales efectos secundarios, que en el caso de la DFC resultarán del todo ineficaces.



