

Dos niñas gemelas fenotípicamente idénticas que nacieron sanas tras un parto normal. Los padres y otros 3 hermanos no presentaban ninguna patología relevante. Las gemelas acuden a nuestra consulta a los 2 años de edad, por presencia de lesiones hiperqueratósicas en las manos y los pies, así como en las regiones periarticulares. No presentaban alteraciones en las mucosas ni en los anejos. Su madre refiere el inicio del cuadro desde los 2 meses de edad, con evolución lenta y progresiva.
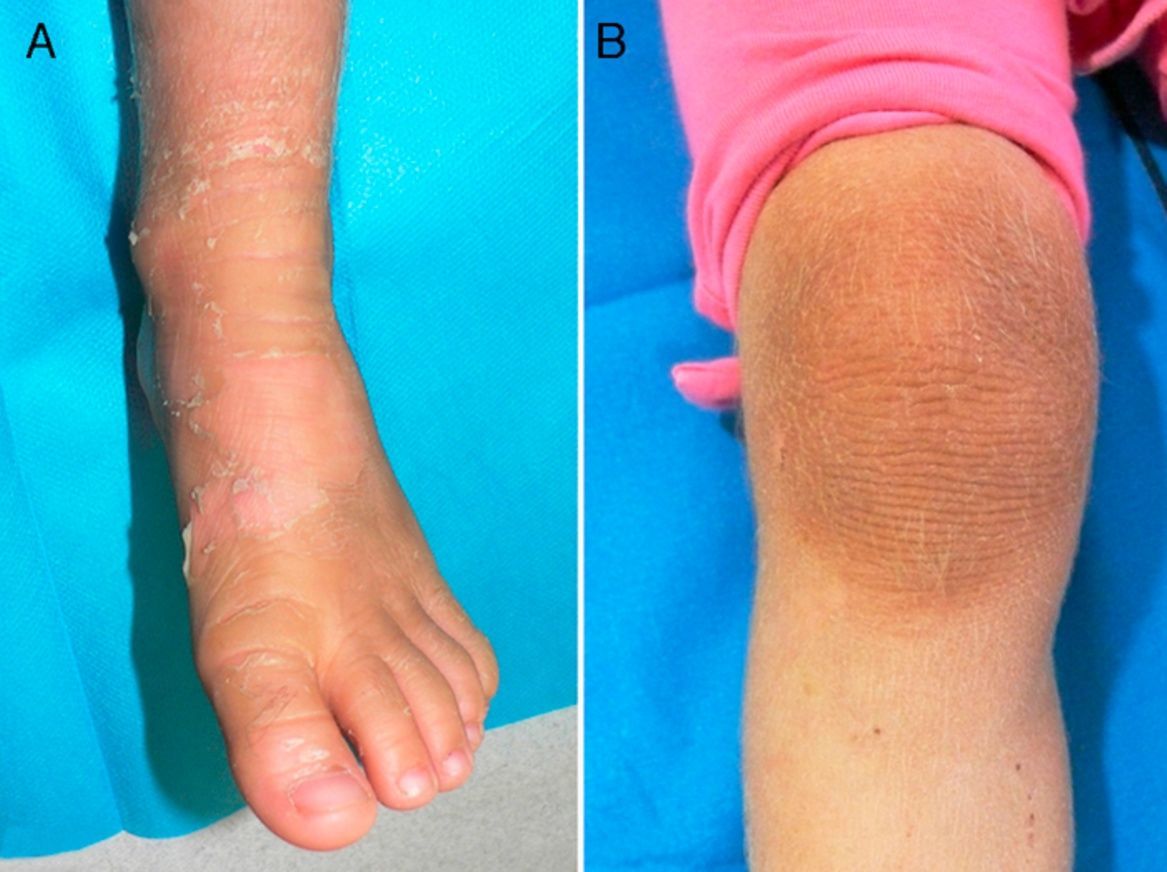
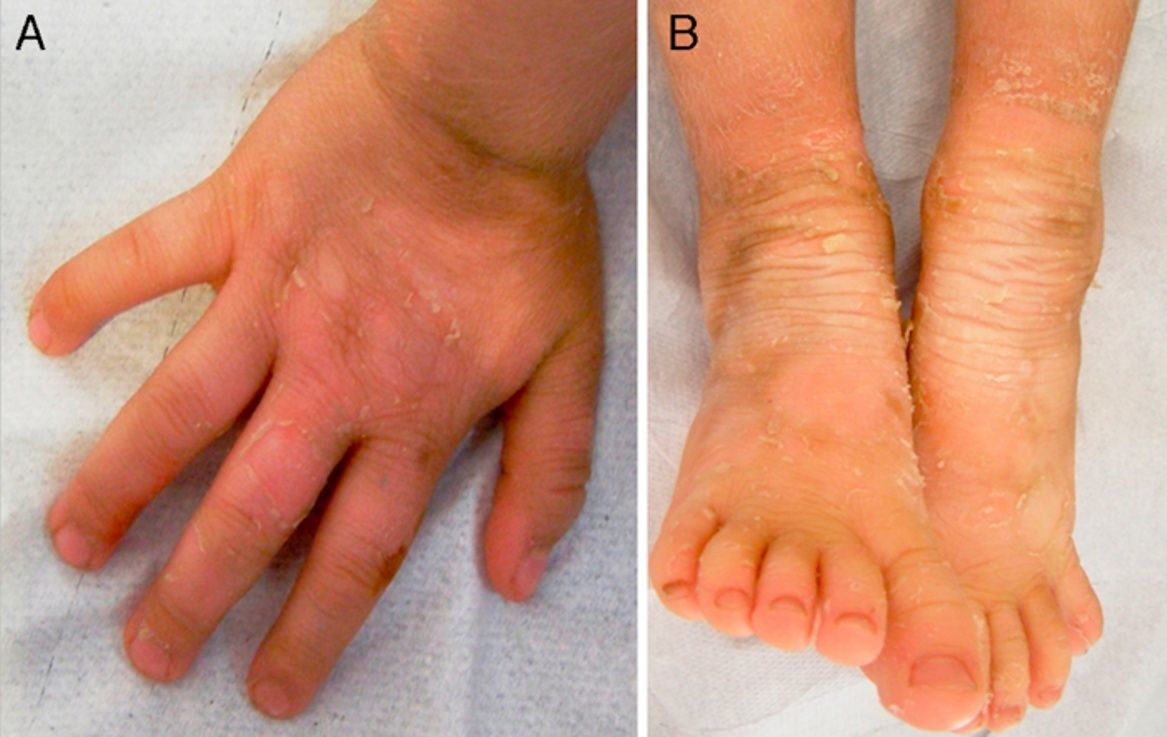
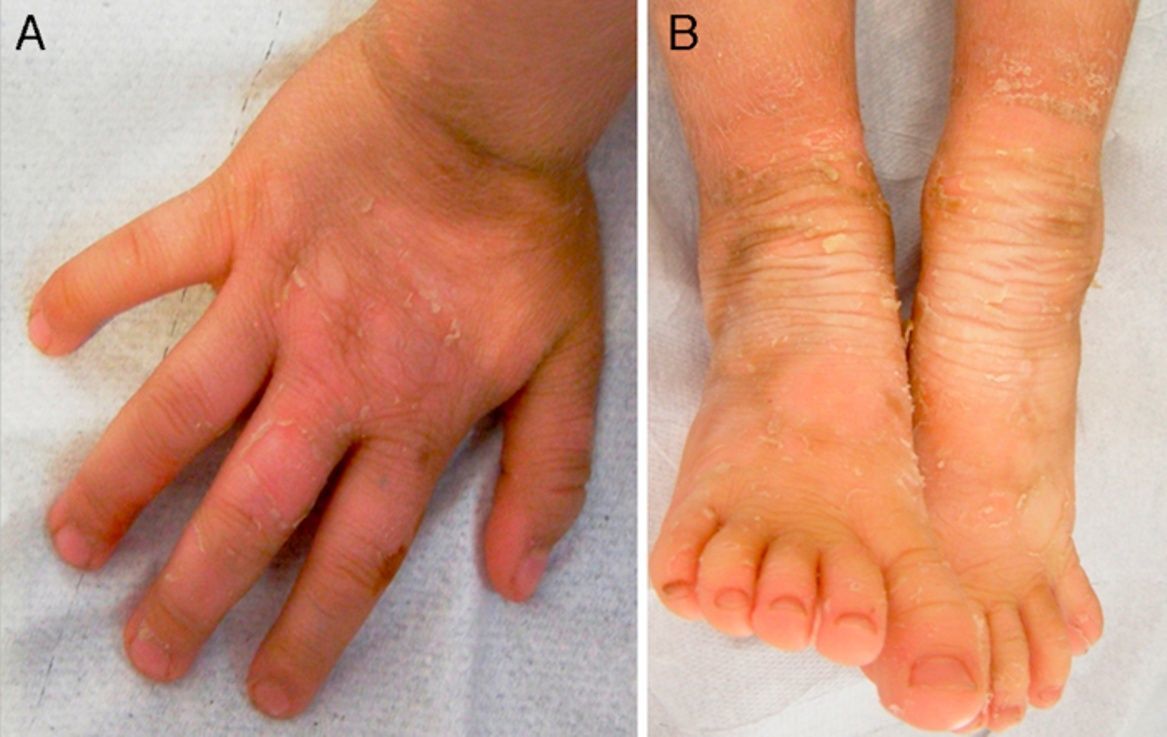
Exploración físicaAmbas pacientes presentaban un cuadro clínico idéntico, con engrosamiento e hiperpigmentación cutánea en los pies y en las manos, en patrón de «guante y calcetín», así como en regiones articulares, con áreas de descamación superficial en grandes láminas. Existía un aumento local de vello fino en las extremidades (fig. 1A y B; fig. 2A y B).
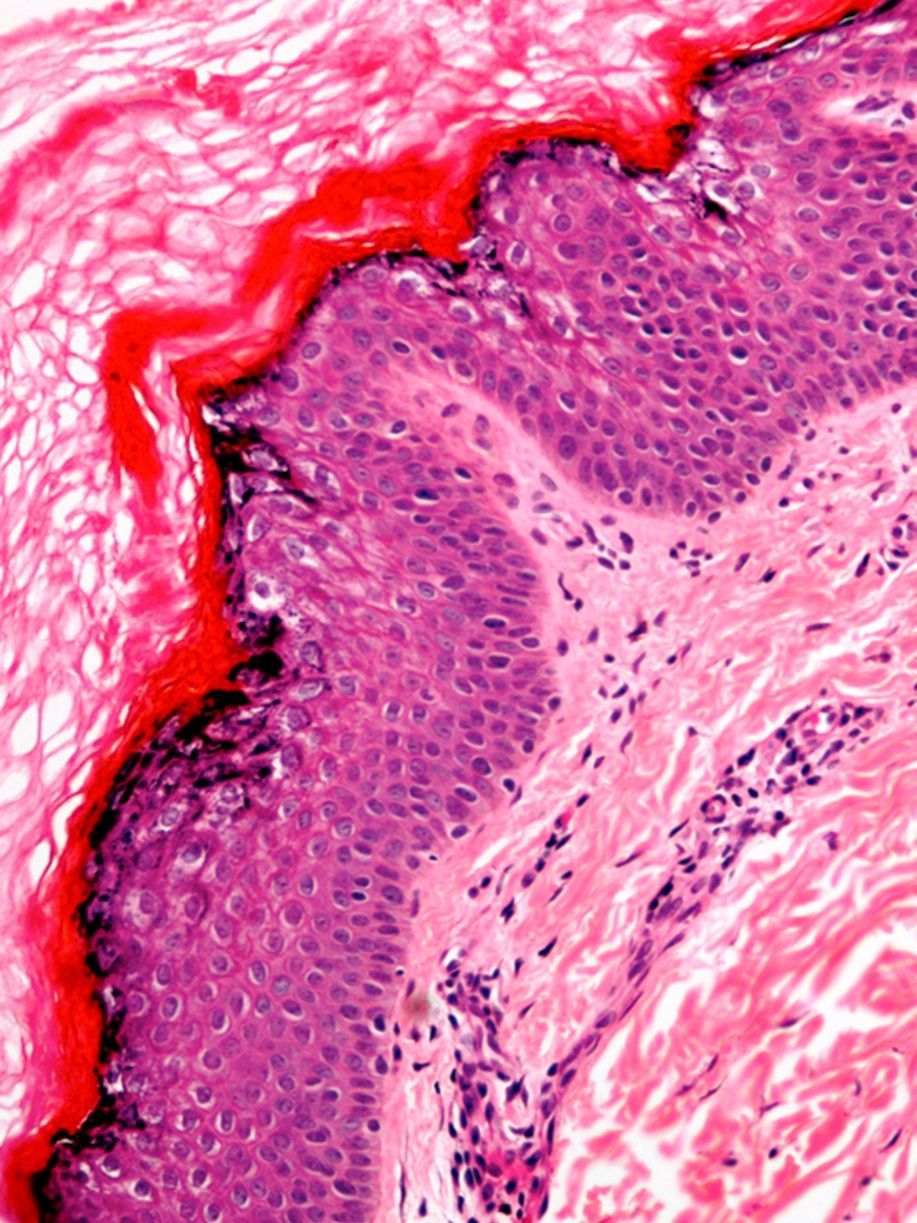
Histopatología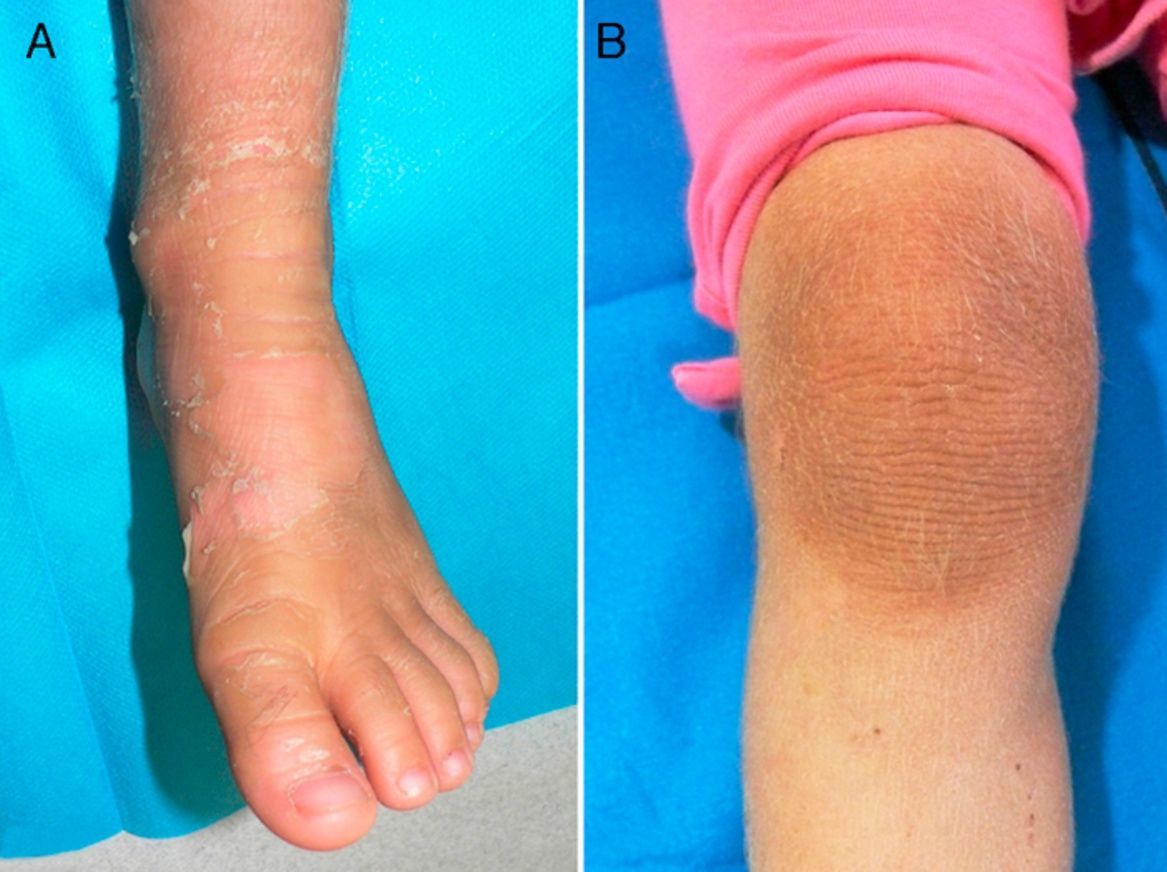
La biopsia cutánea reveló un infiltrado inflamatorio perivascular discreto en la dermis superficial. La epidermis mostraba una hiperqueratosis eosinofílica ortoqueratósica con acantosis epidérmica irregular. El estrato granuloso presentaba una intensa vacuolización de los queratinocitos, con pérdida de las uniones intercelulares y presencia de despegamiento epidérmico. Se observaban gránulos de queratohialina anómalos formando agregados grandes e irregulares (fig. 3).
Otras pruebas complementarias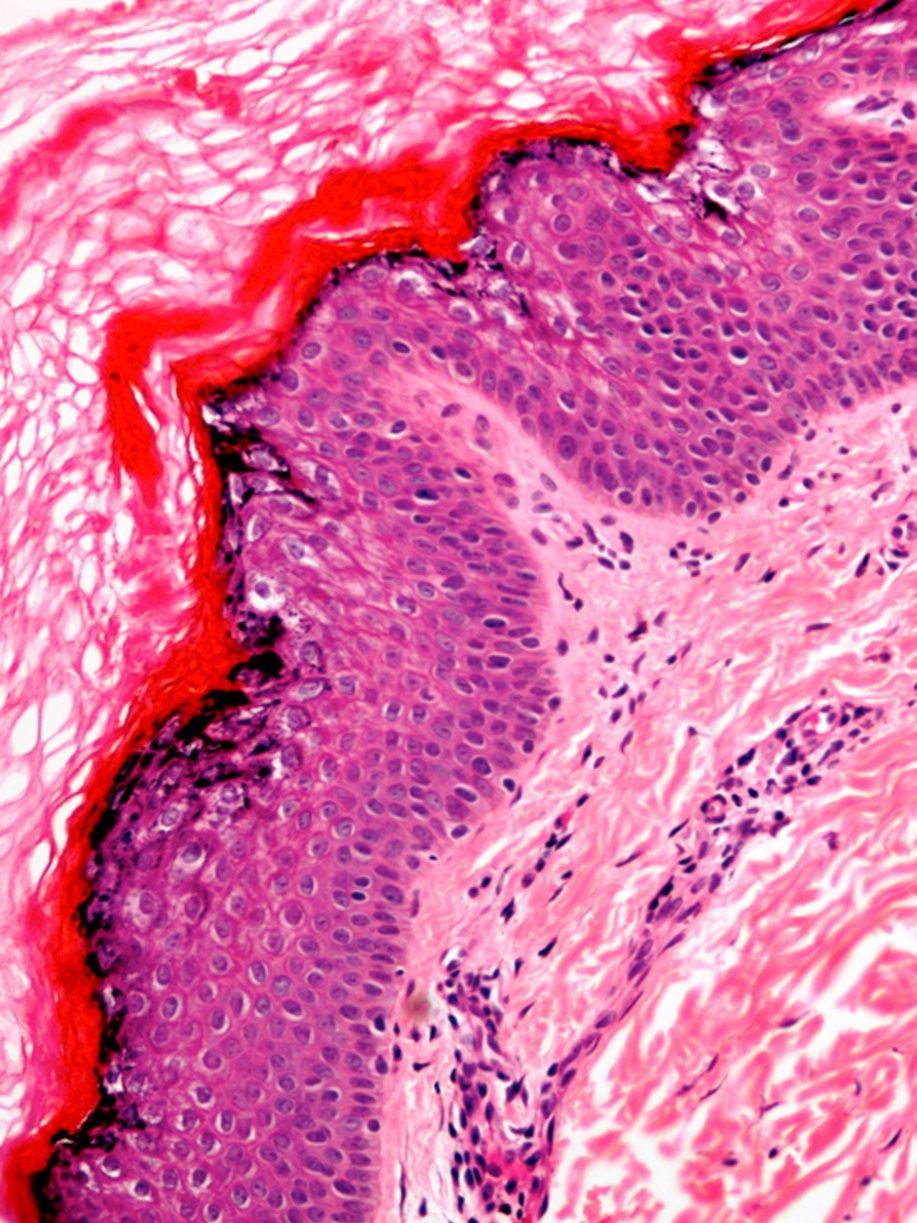
Se realizó secuenciación del gen KRT2 (cromosoma 12q) encontrándose una mutación que produce un cambio de aminoácido en esta proteína.
¿Cuál es su diagnóstico?
DiagnósticoIctiosis bullosa de Siemens (IBS).
Evolución y tratamientoEn ambas pacientes se pautó tratamiento tópico emoliente y tazaroteno al 0,03% tópico con buena respuesta, mejorando las zonas tratadas.
ComentarioLa IBS es un raro trastorno de la queratinización de herencia autosómica dominante (AD) descrito por Siemens en 19371, cayendo en el olvido hasta que Traupe et al. describen una segunda familia afectada en 19862. En 1994 se identificó su origen genético, causado por una mutación de la queratina 2e (KRT2e)3. Macroscópicamente se manifiesta sobre regiones flexurales (rodillas, tobillos, muñecas y codos), en forma de áreas de denudación epidérmica superficial (piel desnuda aparentemente sana) en medio de zonas hiperqueratósicas, signo conocido como «fenómeno de mauserung»4. Este término fue acuñado por Siemens y supone una característica distintiva de importancia clínica, ya que no se presenta en otras formas de ictiosis. Aunque la alteración de KRT2e también se expresa en la región palmoplantar, por motivo desconocido no causa lesiones.
Microscópicamente, la alteración de la KRT2e se expresa en el estrato granuloso y espinoso alto, originando un patrón conocido como hiperqueratosis epidermolítica, caracterizado por una hiperqueratosis compacta y degeneración vacuolar de queratinocitos con alteración de gránulos de queratohialina. Este patrón no es patognomónico, ya que puede encontrarse también en otras entidades como la queratodermia palmoplantar de Vörner o el acantoma epidermolítico, o incluso en la piel sana, donde puede haber focos incidentales de hiperqueratosis epidermolítica. El diagnóstico diferencial principal, tanto clínico como histológico, con el que esta enfermedad es confundida fácilmente, es la eritrodermia ictiosiforme congénita ampollosa (EICA), consistente en alteración de queratinas 1 o 10, que se expresan a nivel más profundo (estrato espinoso suprabasal). A diferencia de la IBS, en la que los niños nacen sanos, en la EICA nacen eritrodérmicos y tienen una clínica más severa con desarrollo de ampollas, descamación extensa y anormalidades hidroelectrolíticas5. El diagnóstico definitivo de la IBS se realiza con el estudio genético. Nuestro caso se trata de gemelas monocigóticas, y por tanto con la misma dotación genética, ambas portadoras de la mutación. No se realizó estudio genético a los padres y hermanos al no tener ninguno de ellos alteraciones fenotípicas. Dado que la IBS es de herencia AD con alta penetrancia, se postula la hipótesis del desarrollo de una mutación espontánea en el cigoto antes de producirse la división del mismo. De este modo, tras su división se transmitiría la mutación a ambas niñas. El tratamiento fundamental es de soporte, con emolientes y queratolíticos tópicos. Una de las complicaciones más frecuentes es el desarrollo de pustulosis sobreañadida por sobreinfección, que requiere el empleo de antibióticos tópicos o sistémicos. La evolución de esta entidad es a la estabilización y la mejoría con la edad, tratándose de una enfermedad relativamente benigna.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Deseamos mostrar nuestro agradecimiento al Dr. Antonio Torrelo por sus útiles aportaciones al diagnóstico.



