









El pénfigo paraneoplásico es una enfermedad ampollosa autoinmune asociada a una neoplasia oculta o previamente diagnosticada, con manifestaciones clínicas, histológicas e inmunológicas bien definidas. Se caracteriza por erosiones de la mucosa orogenital refractarias a los tratamientos convencionales y la presencia de lesiones cutáneas polimorfas. La histología puede ser variada, con presencia de acantólisis o dermatitis liquenoide. La presencia de anticuerpos circulantes es un hallazgo constante que confirma el diagnóstico. Presentamos dos niñas con pénfigo paraneoplásico asociado a linfoma de Hodgkin y enfermedad de Castleman respectivamente. Presentaban compromiso orogenital refractario a tratamientos convencionales. En la histopatología se observó acantólisis y reacción liquenoide respectivamente. Los estudios inmunológicos confirmaron el diagnóstico. Ambas realizaron tratamiento quimioterápico para su enfermedad de base conjuntamente con altas dosis de corticosteroides para sus lesiones cutáneo-mucosas falleciendo por fallo respiratorio. Es de nuestro interés destacar que si bien es infrecuente la presencia de pénfigo paraneoplásico en la edad infantil y en la adolescencia, debe ser tenido en cuenta entre los diagnósticos diferenciales de las dermatosis erosivas periorificiales facilitando, de esta manera, el hallazgo de una neoplasia oculta.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with an occult or previously diagnosed tumor. Its clinical, histological, and immunological features have been clearly defined. It is characterized by the presence of polymorphic skin lesions and by erosions of the oral and genital mucosas that are refractory to conventional treatments. The histology can be variable and includes acantholysis or lichenoid dermatitis. Circulating autoantibodies are a constant feature and confirm the diagnosis. We describe 2 girls with paraneoplastic pemphigus associated with Hodgkin lymphoma in one and Castelman disease in the other. Both children had oral and genital lesions that did not respond to conventional treatments. Biopsy revealed acantholysis in one and a lichenoid reaction in the other, and immunoassays confirmed the diagnosis. Chemotherapeutic treatment of the underlying disease was performed in both cases, together with high-dose corticosteroids for the skin and mucosal lesions. Both patients died due to respiratory failure. We suggest that paraneoplastic pemphigus, although rare in childhood and adolescence, should be included in the differential diagnosis of periorificial erosive dermatitis; this may assist in the detection of an occult neoplasm.
El pénfigo paraneoplásico (PPN), es una enfermedad ampollosa autoinmune que ocurre en presencia de una neoplasia. Es característica la presencia de erosiones dolorosas en las mucosas y polimorfismo cutáneo. Los hallazgos histopatológicos son variables, incluyendo acantólisis y dermatitis de interfase. La presencia de autoanticuerpos dirigidos contra las plaquinas es un hallazgo constante que permite llegar al diagnóstico. El compromiso pulmonar es una de las principales causas de muerte. El PPN es una afección muy poco frecuente en la infancia. En este grupo la neoplasia más frecuentemente asociada es la enfermedad de Castleman.
Presentamos dos pacientes, con linfoma de Hodgkin y enfermedad de Castleman respectivamente, con compromiso mucocutáneo refractario a diferentes tratamientos, cuya clínica y hallazgos inmunopatológicos corresponden al diagnóstico de pénfigo paraneoplásico.
Caso clínico 1Paciente de sexo femenino, 10 años de edad, sin antecedentes personales ni familiares de interés, que comienza 45 días antes de la consulta con lesiones erosivas y ampollas en mucosa oral y anogenital. La paciente se encontraba en mal estado general, con astenia, pérdida de peso e imposibilidad para alimentarse. Al examen físico se observaban erosiones cubiertas con costras hemáticas acompañadas de secreción amarillenta y queilitis en ambos labios y cavidad oral (fig. 1). En los genitales se observaba intenso edema de clítoris y labios mayores, acompañado de grandes erosiones y fisuras anales. Durante su internación la niña presentó un cuadro de dolor torácico agudo y hematemesis.

Se realizaron los siguientes exámenes complementarios:
- 1)
histopatología del labio y mucosa yugal, donde se evidenció una lesión erosionada con pérdida casi total de la epidermis y presencia de fisuras acantolíticas suprabasal y queratinocitos desprendidos por acantólisis, con un denso infiltrado inflamatorio a predominio mononuclear en dermis (fig. 2a). Inmunofluorescencia directa (IFD): IgG=+/++++ rodeando queratinocitos en pequeños focos en toda la epidermis.
- 2)
inmunofluorescencia indirecta (IFI) con esófago de mono y vejiga de rata: se constató que los anticuerpos circulantes marcaban la sustancia intercelular y la membrana basal del epitelio en ambos sustratos (fig. 3).
 en comparación con el testigo, pénfigo vulgar, donde la IgG circulante solamente marca en epitelio de esófago de mono (derecha).") Figura 3.
Figura 3.Caso 1: inmunofluorescencia indirecta. IgG circulante que marca sustancia intercelular y membrana basal del epitelio de esófago de mono y vejiga de rata (izquierda) en comparación con el testigo, pénfigo vulgar, donde la IgG circulante solamente marca en epitelio de esófago de mono (derecha).
(0.28MB). - 3)
inmunoprecipitación: positivo para desmoplaquina i −250kD-, antígeno del penfigoide ampollar −230kD- desmoplaquina ii y envoplaquina −210kD-, periplaquina −190kD- y un antígeno no identificado de 170kD.
- 4)
En una tomografía axial computada (TAC) de tórax se pudo evidenciar una formación sólida, de 7×7×9cm, con áreas hipodensas en su interior y calcificaciones puntiformes, ubicada en mediastino anterior a la izquierda de la línea media, con contorno regular que luego de la inyección de contraste se observó refuerzo parcial con áreas hipodensas, compatibles con necrosis.
- 5)
En la histopatología de la masa tumoral mediastínica se observó una lesión constituída por proliferación difusa de linfocitos maduros, separados por tejido colágeno en variada cantidad, con presencia de células grandes con núcleos voluminosos, hipercromáticos, algunos lobulados con nucléolo evidente, entremezcladas con la población linfoide. La inmunomarcación fue CD20, CD3, PAN T positiva en linfocitos pequeños y fascina positiva en células atípicas, con citoqueratinas, EMA, CD34, CD15 y CD30 negativos. Compatible con linfoma de Hodgkin variedad celularidad mixta con áreas de esclerosis (fig. 2b).
- 6)
Se realizó una esofagoscopía, en donde se observaba úlceras desde la orofaringe hasta el tercio inferior del esófago. En la histopatología, se apreciaba acantólisis a nivel de las células basales con signos de necrosis (fig. 4).
 Biopsia de labio: H-E 20×. Fisuras acantolíticas suprabasales y queratinocitos desprendidos por acantólisis. Infiltrado inflamatorio mononuclear en dermis. b) H-E 20×. Linfoma Hodgkin variedad celularidad mixta con áreas de esclerosis.")
 en comparación con el testigo, pénfigo vulgar, donde la IgG circulante solamente marca en epitelio de esófago de mono (derecha).")

En base a las características clínicas junto con los estudios complementarios y los hallazgos anatomopatológicos, se confirmó el diagnóstico de pénfigo paraneoplásico asociado a linfoma de Hodgkin, estadio ii b 1.
Realizó tratamiento con dos pulsos de metilprednisolona, 30mg/kg/d y dos ciclos de quimioterapia para el linfoma, con un mes de intervalo. El primer ciclo con bleomicina, 15mg/m2 y doxorrubicina, 37,5mg/m2, y el segundo, con vinblastina, 9mg/m2, DTIC, 560mg/m2, bleomicina, 15mg/m2 y doxorrubicina, 37,5mg/m2, con desaparición de la masa tumoral y leve mejoría de sus lesiones cutáneomucosas luego del segundo ciclo de quimioterapia. Durante el mismo, desarrolló un cuadro agudo de insuficiencia respiratoria que requirió asistencia respiratoria mecánica, con mala evolución, falleciendo a los siete meses de haber comenzado su enfermedad.
Caso clínico 2Paciente de 12 años, sin antecedentes personales ni familiares de interés. Comienza cinco meses previos a la consulta, con erosiones y úlceras, dolorosas, en la cavidad oral, mucosa yugal, lengua, labios, mucosa genital y anal sin respuesta a los tratamientos instaurados (fig. 5). Posteriormente presentó dificultad para alimentarse, disminución de peso y mal estado general. A los 3 meses se detectó una tumoración en la región laterocervical izquierda de 9 cm de diámetro, de consistencia duro-pétrea, no dolorosa. Al mes siguiente, presentó lesiones erosivas, dolorosas con fisuras y costras hemáticas en palmas y plantas, con paroniquia y onicodistrofia (fig. 6). Se realizaron los siguientes exámenes complementarios:
- 1)
Una resonancia magnética nuclear (RMN) de cuello, en donde se observaba un conglomerado adenomegálico localizado en la cadena cervical lateral izquierda, de 8×5,3cm de diámetro desplazando las estructuras adyacentes.
- 2)
Una TAC de tórax, en donde se evidenció una imagen nodular con cavitación central en el segmento apical del lóbulo inferior derecho.
- 3)
En la histopatología de palma de la mano se informaba hiperqueratosis ortoqueratótica. Acantosis con alargamiento de los procesos interpapilares; queratinocitos disqueratóticos, espongiosis y vacuolización de los queratinocitos basales con algunos cuerpos apoptóticos En dermis infiltrado inflamatorio linfoplasmocitario en banda en contacto con la capa basal oscureciendo la unión dermo-epidérmica compatible con reacción liquenoide (fig. 7a). La IFD fue negativa.
 H-E 20×. Reacción liquenoide. Queratinocitos disqueratóticos, espongiosis y vacuolización de los queratinocitos basales. Infiltrado inflamatorio linfoplasmocitario en banda. b) H-E 20×. Enfermedad de Castleman tipo hialinovascular. Folículos con centros germinales reactivos prominentes y abundantes histiocitos.") Figura 7.
Figura 7.Caso 2: a) H-E 20×. Reacción liquenoide. Queratinocitos disqueratóticos, espongiosis y vacuolización de los queratinocitos basales. Infiltrado inflamatorio linfoplasmocitario en banda. b) H-E 20×. Enfermedad de Castleman tipo hialinovascular. Folículos con centros germinales reactivos prominentes y abundantes histiocitos.
(0.57MB). - 4)
En el estudio histopatológico de labio y mucosa yugal se observó un epitelio erosionado y estroma subyacente con marcado infiltrado de tipo inflamatorio predominantemente mononuclear y ocasionales células gigantes multinucleadas. Diagnóstico: signos de proceso inflamatorio crónico con cambios sugestivos de pénfigo.
- 5)
En la histopatología de la tumoración se observó un parénquima ganglionar con arquitectura conservada, folículos con centros germinales reactivos prominentes y abundantes histiocitos, en senos y a nivel interfolicular con delgados tabiques fibrosos difusos compatible con hiperplasia ganglionar de tipo enfermedad de Castleman hialinovascular (fig. 7b).
- 6)
La IFI informó anticuerpos antisustancia cementante con sustrato de esófago de mono: positivo +++/++++ y con sustrato de vejiga de rata: negativo.
 H-E 20×. Reacción liquenoide. Queratinocitos disqueratóticos, espongiosis y vacuolización de los queratinocitos basales. Infiltrado inflamatorio linfoplasmocitario en banda. b) H-E 20×. Enfermedad de Castleman tipo hialinovascular. Folículos con centros germinales reactivos prominentes y abundantes histiocitos.")

 Lesiones erosivas y fisuras en palmas de manos. b) Inflamación periungueal con onicodistrofia.")
Realizó 3 ciclos de tratamiento con meprednisona, vía oral, 60mg/m2, junto con rituximab, 375mg/m2, ciclofosfamida, 750mg/m2, vincristina 1,4mg/m2 y doxorrubicina 37,5mg/m2, con intervalo de 21 días. A las 3 semanas de comenzado el tratamiento la niña presentó mejoría de sus lesiones cutáneas, sin modificación de su masa tumoral. A los dos meses de iniciado el mismo comenzó con dificultad respiratoria. Se realizó TAC de tórax en donde se observaba áreas de hipoperfusión en mosaico de distribución difusa bilateral con imagen en vidrio esmerilado con múltiples áreas de consolidación periféricas y leve engrosamiento del intersticio peribroncovascular. En la espirometría se constató una incapacidad obstructiva severa (VEF1 21%). Estos hallazgos fueron compatibles con bronquiolitis obliterante. La niña falleció 2 meses después.
DiscusiónEl pénfigo paraneoplásico es una enfermedad ampollosa autoinmune que se presenta en el contexto de una neoplasia oculta o previamente diagnosticada, descripta por Anhalt et al en 19901–10. Nguyen et al, en el año 2001, propusieron el término de síndrome multiorgánico autoinmune paraneoplásico (PAMS) debido al compromiso de múltiples órganos además del cutáneo y por presentar una patogenia que no se limita a la presencia de autoanticuerpos contra el complejo antigénico conocido. Este síndrome se asocia con mayor frecuencia a desórdenes mieloproliferativos y neoplasias hematológicas tales como linfoma no Hodgkin, leucemia linfocítica crónica, enfermedad de Castleman, timomas, macroglobulinemia de Waldestron, gammapatía monoclonal y linfoma de Hodgkin1–10. En niños y adolescentes, la neoplasia más frecuentemente asociada es la enfermedad de Castleman1–3,6 mientras que en los adultos son los linfomas no Hodgkin y la leucemia linfocítica crónica1,2. Creemos que la discrepancia en la frecuencia de los distintos tumores asociados a PPN en adultos y niños se podría deber a la diferente frecuencia de presentación de dichas neoplasias independientemente de su asociación al PPN. Al igual que lo descrito en la literatura una de nuestras pacientes presentó PPN asociado a enfermedad de Castleman, mientras que en la otra el PPN estaba asociado a linfoma de Hodgkin. Esta última, es una asociación muy poco frecuente, descrita en el 0,6% de los casos2.
El PPN se presenta con mayor frecuencia entre los 45 y 70 años, aunque el rango de edad varía entre los 7 y 83 años2–4,8,9.
El mecanismo fisiopatológico por el cual se producen las lesiones mucocutáneas en el PPN no se conoce con exactitud. Intervienen tanto la inmunidad celular como la humoral. Se han descrito cuatro hipótesis1,3,4,8,10–15:
- 1)
Se postula que los tumores expresan proteínas epiteliales que presentan reacción cruzada con las proteínas de los epitelios del paciente. Al producirse una respuesta inmune antitumoral, también se afectarían los epitelios produciendo las lesiones cutáneo-mucosas. Zhangs et al demostraron que diferentes epitopes presentes en las plaquinas de los epitelios se detectaron en la enfermedad de Castleman y en timomas11.
- 2)
Se propone que existiría una disregulación del sistema inmune producida por la neoplasia, como en el caso de los timomas, que podría producir un autoanticuerpo de tipo pénfigo contra los componentes de los desmosomas y hemidesmosomas. Además se ha demostrado la presencia de niveles aumentados de IL6, secretados por las células tumorales, como lo observado en los linfomas no Hodgkin, leucemia linfocítica crónica y enfermedad de Castleman, lo cual promueve la diferenciación de células B y la producción de inmunoglobulinas. Los niveles elevados de IL6 se relacionan con ciertos procesos autoinmunes como la miastenia gravis y las citopenias autoinmunes. Cuando se extirpan los tumores involucrados los síntomas de las enfermedades autoinmunes desaparecen, lo cual indica que la alteración del sistema inmune juega un rol importante.
- 3)
Nguyen et al12 demostraron la presencia de linfocitos T citotóxicos, macrófagos y células natural killer (NK) en los tejidos involucrados en el PPN, avalando la intervención de la inmunidad celular en la fisiopatología de esta entidad. Se ha demostrado que las células NK y macrófagos inducen una respuesta citotóxica en las células tumorales sin especificidad antigénica; mientras que los linfocitos T citotóxicos reconocen moléculas del complejo mayor de histocompatibilidad (CMH) con fragmentos de péptidos antigénicos expresados en la superficie celular de las células tumorales. Esta asociación, de respuesta humoral y citotóxica es también observada en las enfermedades autoinmunes14.
Reich et al demostraron en un paciente con PAMS la acumulación selectiva a nivel de la epidermis de linfocitos CD8 junto al aumento de la producción de interferàn α y del factor de necrosis tumoral α, con una importante expresión del antígeno DR en los leucocitos. La apoptosis sería el mecanismo clave por el cual se produciría la muerte de los queratinocitos14. Cummins et al presentaron en una serie de casos 4 pacientes con una variante liquenoide de PPN en donde no se observaban autoanticuerpos, concluyendo que en algunos casos de PPN, la inmunidad celular, más que los autoanticuerpos, tendría un papel predominante o exclusivo en la fisiopatología de esta enfermedad15.
- 4)
La teoría de «Epitope spreading» o dispersión de epitopes. Este fenómeno se define como el mecanismo por el cual una enfermedad autoinmune puede producir daño tisular de manera tal que ciertos epitopes previamente escondidos queden expuestos y así generen una respuesta autoinmune. Este mecanismo estaría involucrado en aquellos casos en que el PPN se presente en un inicio como una reacción liquenoide4,11. Nguyen et al proponen que la óxido nítrico sintetasa presenta un papel importante en este proceso. Se comprobó que en las lesiones de esta enfermedad existen queratinocitos con óxido nítrico sintetasa en la epidermis y que estos podrían ser blanco de linfocitos CD8+ y CD68+ que inducirían a la muerte celular programada a través de la liberación de óxido nítrico. De esta forma quedarían expuestos los autoantígenos previamente escondidos al sistema inmune12,14.
Un hallazgo constante, y primer signo de la enfermedad, es el compromiso de la mucosa oral. Es característica la presencia de erosiones y ulceraciones dolorosas que afectan la mucosa y se extienden hasta el bermellón de los labios, de muy difícil manejo y refractarias al tratamiento. Otras mucosas, como la conjuntival, esofágica y anogenital también pueden estar comprometidas1–8,10. Las lesiones cutáneas son muy variadas. Se caracterizan por la presencia de ampollas fláccidas o tensas, con o sin erosiones y/o lesiones de tipo eritema multiforme, liquen plano o injerto contra huésped1–10. Este polimorfismo lesional puede estar presente en un mismo paciente en un determinado momento o a lo largo de la evolución de la enfermedad3. En los niños es más frecuente encontrar una dermatitis liquenoide en tronco y miembros, que puede extenderse hacia la cara y el cuello1,2. Es habitual el compromiso palmoplantar y la afectación ungueal1–8,10. Nuestras pacientes comenzaron con compromiso de la mucosa oral y genital asociado a lesiones palmo-plantares en una de ellas.
El árbol traqueobronquial puede estar directamente comprometido, produciendo un fallo respiratorio por la presencia de bronquiolitis obliterante debido a los autoanticuerpos contra las plaquinas del epitelio bronquial, que serían los responsables de los cambios acantolíticos en el mismo3–5,7,10. Está presente en el 30% de los casos y puede desarrollarse al mes o hasta un año posterior al diagnóstico. Debemos sospecharla ante la presencia de dificultad respiratoria progresiva. Creemos que la primera niña presentó esta complicación, pero no lo pudimos confirmar por la rápida y desfavorable evolución. En la otra paciente se realizó el diagnóstico de bronquiolitis obliterante a través de pruebas de la función pulmonar y TAC de tórax, evidenciándose los hallazgos característicos.
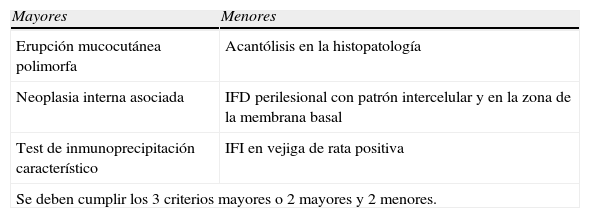
El diagnóstico de PPN se realiza a través de la observación clínica, de los hallazgos histológicos y de los estudios inmunológicos. Basados en los criterios descriptos por Anhalt et al en 1990 (tabla 1), revisados por Camisa y Helm, quienes los dividieron en mayores y menores (tabla 2)1–12, se deben cumplir tres criterios mayores o dos mayores y dos menores.
Criterios de Anhalt et al
|
Modificado de Olguin MF, Pénfigo paraneoplásico. Dermatol Argent 2009;15(2):97–105.
Criterios diagnósticos de Camisa y Helm
| Mayores | Menores |
| Erupción mucocutánea polimorfa | Acantólisis en la histopatología |
| Neoplasia interna asociada | IFD perilesional con patrón intercelular y en la zona de la membrana basal |
| Test de inmunoprecipitación característico | IFI en vejiga de rata positiva |
| Se deben cumplir los 3 criterios mayores o 2 mayores y 2 menores. | |
Modificado de Kimyai-Asadi A, Jih MH. Paraneoplastic pemphigus. Int J Dermatol 2001;40(6):367–72.
Los hallazgos histológicos son tan variados como el polimorfismo lesional que presentan estos pacientes. Se puede observar acantólisis intraepidérmica y/o dermatitis liquenoide con diferentes grados de necrosis de los queratinocitos. Esto se debe a la coexistencia de una respuesta humoral y celular producto de un fenómeno de autoinmunidad contra el epitelio1,3,4,9. En los menores de 20 años, se observa con mayor frecuencia un patrón liquenoide1, tal como se describe en el segundo caso.
Dentro de los estudios inmunológicos que ayudan a confirmar el diagnóstico, podemos detallar:
- 1.
Inmunofluorescencia directa: en el área perilesional se observan depósitos de IgG con o sin complemento interqueratinocítico, en forma linear o granular. Además, es frecuente identificar depósitos de IgG y/o complemento en la zona de la membrana basal. Estos últimos ayudan a diferenciar el pénfigo paraneoplásico de otros pénfigos en donde por lo general los depósitos de inmunoglobulinas son interqueratinocitos y no en la membrana basal3,4,12. Es importante remarcar que existe un número considerable de falsos negativos y que muchas veces se requieren múltiples biopsias para establecer el diagnóstico. Esto se puede deber a los diferentes mecanismos fisiopatológicos de los distintos tipos de lesiones, o por el hecho de que muchas veces solo se observa tejido necrótico en la biopsia de las mucosas1,3. En los pacientes con lesiones de tipo liquenoide la eIFD suele ser negativa, demostrando, en estos casos, la predominancia de la inmunidad celular sobre la humoral7,13,15 como pudimos constatar en nuestra segunda paciente que presentaba una variante liquenoide de PPN con IFD negativa.
- 2.
Inmunofluorescencia indirecta: los pacientes con PPN poseen anticuerpos en suero que reaccionan contra las desmoplaquinas presentes en todos los epitelios (epitelio estratificado de esófago de mono y epitelio transicional de vejiga de rata), mientras que los pacientes con otro tipo de pénfigo presentan anticuerpos que reaccionan contra antígenos presentes únicamente en los epitelios estratificados (desmogleína 1 en el pénfigo foliáceo y desmogleína 3 en el vulgar). Esto ha llevado a considerar este método altamente sensible y específico. Sin embargo Anhalt et al, han demostrado en un estudio con 28 pacientes, que la sensibilidad de esta técnica es del 75% y la especificidad del 83%. Esto se debe a que no todos los pacientes con PPN poseen los anticuerpos dirigidos contra todos los antígenos del complejo. Los que no posean anticuerpos antidesmoplaquinas, pero sí envoplaquina, periplaquina y desmogleínas, no mostrarán fluorescencia en el epitelio transicional de vejiga de rata ya que estos últimos no se encuentran en dicho epitelio3,4,12.
- 3.
Inmunoprecipitación: es el principal estudio utilizado para confirmar el diagnóstico. Se observan: desmoplaquina i −250kD- antígeno de penfigoide ampollar −230kD- desmoplaquina ii y envoplaquina −210kD- periplaquina −190kD- y un antígeno no identificado de 170kD3,4,10,12.
En la primera paciente se cumplieron los criterios mayores y dos de los menores. En la segunda se comprobaron dos criterios mayores sin poder realizar la inmunoprecipitación, dado que no está disponible en nuestro país, y un criterio menor, con una IFI en esófago de mono positiva pero en vejiga de rata negativa. Esto se podría deber, como fue explicado anteriormente en inmunofluorescencia indirecta, a que no todos los pacientes presentan los anticuerpos dirigidos contra todos los antígenos del complejo y los que no tengan antidesmoplaquinas no mostrarán fluorescencia en dicho epitelio3,4,12.
Dentro de los diagnósticos diferenciales debemos tener en cuenta las patologías que cursen con estomatitis refractarias, entre ellos el eritema multiforme, síndrome de Steven Johnson, necrólisis epidérmica tóxica, pénfigo vulgar, penfigoide ampolloso, liquen plano, dermatitis por radiación e infecciones micóticas o herpéticas3,4. El tratamiento de esta enfermedad puede dividirse en dos categorías, por un lado el manejo de la neoplasia y por otro el fenómeno autoinmune. La mayoría de los pacientes con afecciones benignas, mejoran o se curan luego de la extirpación quirúrgica. En el caso de una neoplasia maligna, no existe una terapéutica eficaz, y a pesar de la extirpación quirúrgica de la misma y/o del uso de quimioterapia, esto no impide la progresión de la enfermedad4. En aquellos casos que se presentan con bronquiolitis obliterante el pronóstico es reservado2,5,6. Para el PPN, los corticoides a altas dosis son el tratamiento de elección. El uso concomitante de drogas inmunosupresoras reduciría la dosis de esteroides limitando así los efectos adversos de los mismos. La combinación más frecuentemente utilizada es meprednisona, 1–2mg/kg/d y ciclosporina A, 5mg/kg/d4–10. Sin embargo, no se han obtenido resultados favorables. Tampoco el uso de plasmaféresis, gamma globulina endovenosa, dapsona ni sales de oro ha demostrado ser efectivos5,7. Se han publicado resultados prometedores con el uso de rituximab, anticuerpo monoclonal anti CD20, en los casos de PPN y linfoma de células B2,5,6.
El pénfigo paraneoplásico presenta una tasa de mortalidad del 90%. Las causas de muerte suelen ser las complicaciones de la enfermedad y/o del tratamiento: sepsis, falla multiorgánica, hemorragias gastrointestinales o fallo respiratorio, producto de la bronquiolitis obliterante.1–10
ConclusiónEl PPN, entidad excepcional antes de los 20 años de edad, es un desorden con características clínicas e histopatológicas bien definidas asociado a una neoplasia linfoproliferativa benigna o maligna. En niños y adolescentes predomina un patrón liquenoide asociado, en la mayoría de los casos, a la enfermedad de Castleman. La respuesta al tratamiento depende del tipo de neoplasia y la presencia de complicaciones. La bronquiolitis obliterante es una complicación frecuente y mortal. La denominación de síndrome multiorgánico autoinmune paraneoplásico se debe a que se ha demostrado el compromiso de múltiples órganos, preferentemente el pulmonar, que constituye muchas veces la causa de fallecimiento, y la presencia de autoanticuerpos que no se limitan al complejo antigénico conocido, como la presencia de anticuerpos contra la plectina, antidesmogleína 1 y 3. El diagnóstico precoz facilitará el hallazgo de la neoplasia oculta, en los casos que no se haya detectado, como así también la implementación temprana de un tratamiento adecuado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



