
















INTRODUCCIÓN
El término linfoma cutáneo primario define aquellos linfomas que se desarrollan en la piel sin evidencia de afectación extracutánea en el momento del diagnóstico tras una estadificación completa que incluya un estudio de médula ósea y exploraciones radiológicas. La excepción a esta regla la constituyen, dentro de los linfomas cutáneos de células T, el grupo de la micosis fungoide/síndrome de Sézary, que puede considerarse siempre como linfomas cutáneos primarios, aunque la diseminación extracutánea se haya producido antes de establecer el diagnóstico. En 1997, la European Organization for Research and Treatment of Cancer (EORTC) Lymphoma Program Project Group propuso una clasificación específica para los linfomas cutáneos primarios1. Dicha clasificación define junto a aspectos clínicos y de comportamiento biológico, las características histopatológicas, inmunofenotípicas y genéticas de los diferentes subtipos de linfomas cutáneos. Más recientemente, se ha establecido la nueva clasificación de las neoplasias hematológicas por la Organización Mundial de la Salud (OMS)2-4, que ha incorporado los linfomas cutáneos primarios. Sin embargo, dado su carácter eminentemente práctico, la clasificación de la EORTC continúa siendo la más utilizada por los dermatólogos en el manejo de los linfomas cutáneos (tabla 1)5.
METODOLOGÍA DIAGNÓSTICA EN LOS LINFOMAS CUTÁNEOS DE CÉLULAS T
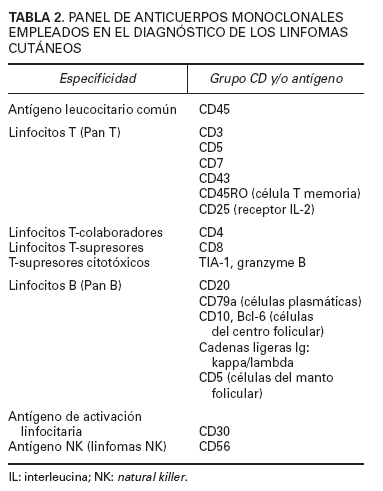
En el diagnóstico de los linfomas cutáneos primarios resulta fundamental establecer una correlación entre la información clínica y la histopatología. Las manifestaciones clínicas pueden permitir establecer la diferenciación entre procesos con una imagen histopatológica similar (papulosis linfomatoide/linfoma cutáneo CD30+) o, incluso, inducir una sospecha diagnóstica en el caso de lesiones precoces con hallazgos histopatológicos no específicos (micosis fungoide). La obtención de una biopsia representativa resulta esencial para valorar las características arquitecturales de estas entidades, por lo que suelen recomendarse biopsias amplias (biopsias en huso) y, en determinadas circunstancias, biopsias repetidas para establecer un diagnóstico definitivo. El estudio histotoplasmáticos permite caracterizar las poblaciones celulares linfoides neoplásicas y acompañantes ya sean de origen T, o B. La pérdida de antígenos linfoides maduros (CD7, CD2), así como la expresión de marcadores de activación linfoide pueden indicar una progresión hacia un proceso linfoproliferativo maligno; es decir, la expresión de un patrón anormal de antígenos linfoides de células T sugeriría el diagnóstico de linfoma cutáneo de células T y no de un proceso linfoide reactivo (tabla 2).

TABLA 1. CLASIFICACIÓN DE LINFOMAS CUTÁNEOS
DE CÉLULAS T PRIMARIAS
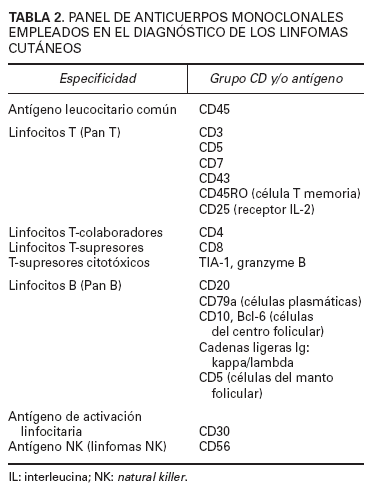
TABLA 2. PANEL DE ANTICUERPOS MONOCLONALES
EMPLEADOS EN EL DIAGNÓSTICO DE LOS LINFOMAS
CUTÁNEOS
Finalmente, la demostración de una proliferación linfoide monoclonal en casos en los que se sospecha el diagnóstico de un linfoma puede realizarse a través del estudio del ADN obtenido a partir de biopsias cutáneas para valorar el reordenamiento de los genes del receptor de la célula T (RCT). Este análisis genotípico comienza a hacerse de forma rutinaria bien mediante Southern blot de la cadena , o más habitualmente mediante la técnica de reacción en cadena de la polimerasa (PCR) de la cadena (o ) del receptor de la célula T. La mayor comodidad, sensibilidad, rapidez, su menor coste y la posibilidad de utilizar material de archivo hacen que esta última técnica sea la más comúnmente empleada. La interpretación de los resultados del análisis geno-típico debe realizarse siempre dentro del contexto de las características clínicas, histológicas e inmunofenotípicas de cada caso. Los resultados falsamente positivos no son excepcionales y, además, existen diversos ejemplos de enfermedades con comportamiento clínico benigno en las que se detectan reordenamientos linfoides monoclonales6,7.
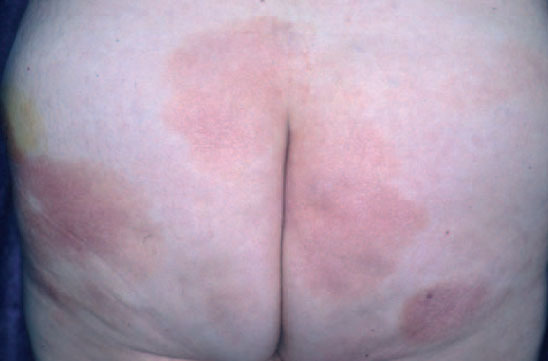
Fig. 1.—Lesiones maculares iniciales de la micosis fungoide.
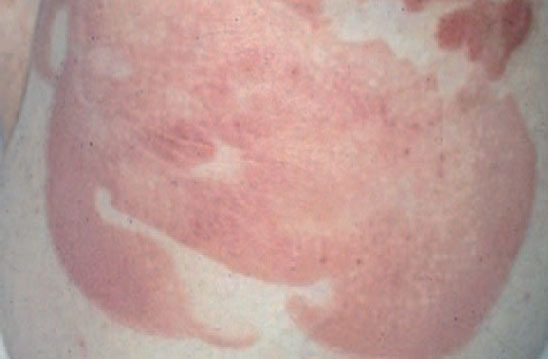
Fig. 2.—Lesiones tipo placa infiltrada en la micosis fungoide.
MICOSIS FUNGOIDE
DiagnósticoAspectos clinicopatológicos
La micosis fungoide se considera un linfoma indolente originado de los linfocitos CD4+ recirculantes que presentan una intensa afinidad para la piel y especialmente la epidermis (epidermotropismo). Es un proceso linfoproliferativo que suele aparecer por regla general en la edad adulta.
La micosis fungoide suele iniciarse como máculas eritematosas persistentes y descamativas, que suelen plantear el diagnóstico diferencial con eczemas o con procesos inflamatorios poco específicos (fig. 1). En la fase de placas las lesiones se hacen de forma gradual más sobreelevadas e infiltradas al tacto. Dichas lesiones pueden desarrollarse a partir de lesiones maculares o aparecer de novo sobre piel previamente normal (fig. 2). Las placas varían en forma y tamaño, pueden regresar espontáneamente, permanecer estables, o evolucionar a la formación de nódulos o tumores (fig. 3). De forma similar, los tumores pueden desarrollarse a partir de placas preexistentes o a partir de una eritrodermia, o pueden originarse a partir de piel normal. Con frecuencia tanto las placas como los tumores tienden a presentar necrosis y ulceración. Otra posible manifestación clínica de la micosis fungoide es una eritrodermia exfoliativa. Rara vez la formación de tumores está presente ya en el inicio de la enfermedad. La evolución hacia eritrodermia puede producirse en cualquier momento de la enfermedad y su distinción de un síndrome de Sézary dependerá de si se ajusta o no a los criterios diagnósticos de este último proceso. En la fase macular o de placas puede observarse la presencia de linfadenopatías reactivas superficiales (linfadenopatía dermatopática), mientras que en las fases avanzadas (estadio tumoral) pueden palparse linfadenopatías profundas específicas, a veces asociadas a lesiones viscerales (esplénicas, pulmonares, gastrointestinales, hepáticas, etc.)8.
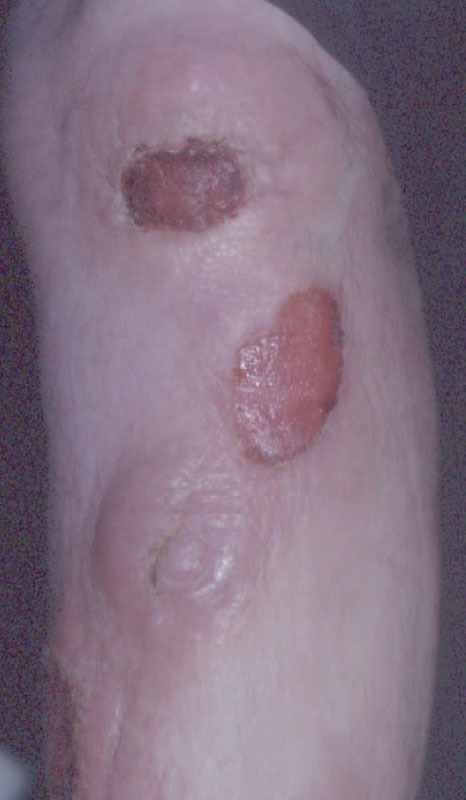
Fig. 3.—Lesiones tumorales de fases avanzadas de la micosis fungoide.
La reticulosis pagetoide es una variante localizada de micosis fungoide que suele localizarse en las zonas distales de las extremidades y se caracteriza por un intenso epidermotropismo con escaso infiltrado dérmico subyacente9. La denominada piel laxa granulomatosa (granulomatous slack skin) es una peculiar variante de micosis fungoide caracterizada clínicamente por la formación paulatina de piel redundante y laxa en flexuras y, desde un punto de vista histopatológico, por una reacción inflamatoria granulomatosa con células T atípicas y fenómenos de destrucción de las fibras elásticas (elastolisis). El fenotipo de los linfocitos es T cooperador y puede detectarse clonalidad mediante el reordenamiento del RCT por PCR. En una tercera parte de los casos puede asociarse a linfoma de Hodgkin y ocasionalmente a verdadera micosis fungoide. Suele presentar una evolución indolente. La micosis fungoide asociada a mucinosis folicular es otra variante clínica caracterizada por una intensa infiltración de los folículos pilosos con una degeneración mucinosa del epitelio folicular. Desde un punto de vista clínico se manifiesta en forma de pápulas foliculares, placas o tumores que suelen localizarse en la región de la cabeza y el cuello. Puede asociarse con alopecia (alopecia mucinosa) y prurito. Otras variantes clinicopatológicas raras cuyo conocimiento es interesante para un diagnóstico adecuado incluyen la micosis foliculotropa (no asociada a mucinosis folicular), siringotropa, granulomatosa, tipo pitiriasis liquenoide, ictiosiforme, verrugosa, purpúrica, hipopigmentada, pustulosa, vesiculosa ampollosa e «invisible». En todas ellas se encuentran rasgos histopatológicos de micosis fungoide que determinan el diagnóstico7,10-12.
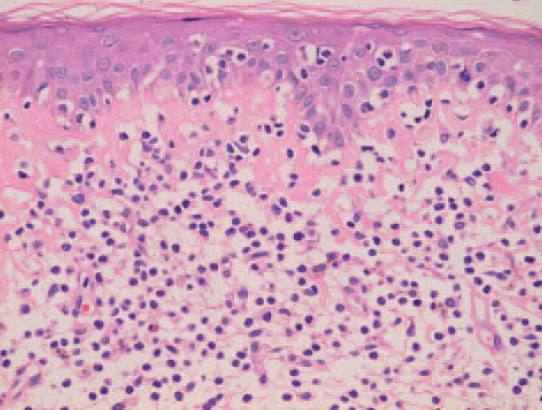
Fig. 4.—Epidermotropismo de células cerebriformes en lesión incipiente de micosis fungoide. (Hematoxilina-eosina, ×200.)
El estudio histopatológico evidencia unos cambios morfológicos distintos según las características clínicas de las lesiones. En las lesiones iniciales los cambios observados pueden no ser diagnósticos y requerir la práctica de numerosas biopsias. En las lesiones más avanzadas se observa un infiltrado en banda formado por células mononucleares de tamaño pequeño o intermedio con núcleo cerebriforme hipercromático sin espongiosis. De forma característica se observa epidermotropismo de los linfocitos cerebriformes atípicos, tanto en forma de células aisladas o formando agregados, los denominados microabscesos de Pautrier (fig. 4). En los estadios tumorales, las biopsias cutáneas muestran un infiltrado monomorfo formado por células atípicas de gran tamaño con frecuentes mitosis que suele afectar a todo el espesor de la dermis. En estas fases tardías no suele observarse epider-motropismo13,14.
Aspectos inmunofenotípicos y genéticos
El fenotipo predominante es CD3/CD4+ cooperador aunque algunos casos expresan un fenotipo CD3/CD8+ supresor/citotóxico presentando estos últimos un curso por lo general más agresivo. También el fenotipo citotóxico es el predominante en aquellos casos de reticulosis pagetoide. La mayor parte expresan antígenos pan-T como CD3 y CD5 así como CD2, pero no es infrecuente la pérdida de alguno de ellos (CD5, CD7, CD2) hasta en el 50-75 %, sobre todo en las formas tumorales15. Los estudios de reordenamiento demuestran clonalidad en la mayoría de los casos (80 %) y al menos en el 50 % de los pacientes en estadios iniciales. Ocasionalmente se han detectado anomalías citogenéticas, aunque no específicas, como por ejemplo mutaciones, deleciones o silenciamiento (metilación) de genes reguladores del ciclo celular (p53, p16 o p15)16-18.
La micosis fungoide en estadios precoces (mácula o placa) suele diagnosticarse por correlación clinicopatológica, ya que los hallazgos inmunohistoquímicos a menudo no son concluyentes. En ocasiones puede ser muy difícil establecer el diagnóstico diferencial de una dermatosis inflamatoria crónica como eczema de contacto o neurodermatitis, requiriendo sucesivas biopsias hasta establecer un diagnóstico definitivo. Las lesiones tumorales de micosis fungoide plantean el diagnóstico diferencial clínico con lesiones de leucemia cutis, metástasis cutáneas, o un linfoma cutáneo CD30+ o pleomórfico de células T. En pacientes con una micosis fungoide eritrodérmica o con un síndrome de Sézary, el diagnóstico diferencial debe plantearse con diversas enfermedades que causan una eritrodermia, como dermatitis atópica, toxicodermias y psoriasis.
Estadificación y aspectos pronósticos
La valoración de un paciente con micosis fungoide y el estudio de extensión de la enfermedad debe incluir una historia clínica y una exploración física completas minuciosas, con especial atención a la presencia de adenopatías o de hepatosplenomegalia. Las exploraciones complementarias necesarias incluyen una bioquímica general y hepática, además de un hemograma completo con citología de sangre periférica (recuento manual e inmunofenotipo mediante citometría de flujo), radiografía de tórax, tomografía computarizada (TC) toracoabdominal y pélvica, y biopsia ganglionar en caso de adenopatía palpable. No se recomienda la práctica rutinaria de un estudio medular (biopsia de médula ósea) a excepción de los pacientes con estadios tumorales, en casos con una elevada sospecha de enfermedad visceral o en pacientes con fenotipo citotóxico (CD8+, granzyme, TIA-1), o con receptor de la célula T de fenotipo gamma/delta. El estudio del re-ordenamiento genético del receptor de la célula T es opcional en caso de un diagnóstico dudoso19 (tabla 3).

TABLA 3. ALGORITMO DIAGNÓSTICO DE LINFOMA CUTÁNEO DE CÉLULAS T
Los estadios de la enfermedad se definen por la clasificación TNM. Los factores pronósticos más importantes en un estudio multivariable son la presencia de enfermedad visceral y el tipo de lesión cutánea según el estadio (tabla 4). Algunos estudios sugieren que los pacientes pueden dividirse en tres categorías: a) estadios IA, IB, IIA, con excelente tasa de supervivencia (estadio IA una supervivencia media superior a 12 años; pacientes con estadio IB y IIA con una esperanza de vida a los 5 años en torno al 75-85 % y 50-70 %, respectivamente); b) estadios IIB-III con supervivencia intermedia (40 % a los 5 años), y c) un grupo de alto riesgo, estadios IVA-IVB con una supervivencia pobre (menor a 2,5 años o 0-15 % a los 5 años)20,21.
La presencia de un porcentaje elevado de linfocitos CD8+ en el infiltrado tumoral en mayor proporción parece ser indicador de mejor pronóstico. En los estadios iniciales de la enfermedad la inmunidad celular sigue un patrón similar al individuo normal con predominio de respuesta celular citotóxica (NK, CD8+) y patrón de citocinas Th1 (interferón gamma [IFN-α], interleucinas [IL-2, IL-12]).
Se desconoce el valor pronóstico real de la presencia de un clon en sangre periférica, pero algunos estudios sugieren que aquellos pacientes con micosis fungoide en estadio precoz en los que este se demuestra tienen más probabilidad de recaídas22. La transformación citológica de un linfoma de bajo grado a uno de alto grado histológico (con al menos el 25 % de células grandes) se asocia con un peor pronóstico. La presencia de mucinosis folicular y el grosor del infiltrado dérmico también son otros elementos de pronóstico desfavorable en algunos estudios23.
Aunque la mayor parte de los pacientes no progresan o permanecen en un estadio precoz con lesiones en placas, en alrededor del 10 % de los casos se produce la progresión a enfermedad avanzada cutánea (tumor) y extracutánea (ganglios linfáticos, vísceras, sangre). Sólo una minoría de los pacientes con micosis fungoide fallece por su enfermedad.
TratamientoEl tratamiento de la micosis fungoide suele depender de las características clínicas y de la estadificación de la enfermedad. Los distintos abordajes terapéuticos disponibles consiguen únicamente una mejoría sintomática y clínica (remisión completa o parcial). Sin embargo, no existe ningún tratamiento definitivo o curativo de la enfermedad. Las remisiones completas son frecuentes, especialmente en estadios precoces (tabla 5). La prescripción de estrategias terapéuticas agresivas en estadios precoces no ha demostrado obtener una mejoría con respecto a alcanzar un mayor tiempo libre de enfermedad, ni un aumento con respecto a la supervivencia global. En el único estudio clínico aleatorizado que comparaba en un grupo de 103 pacientes con micosis fungoide en distintos estadios la eficacia de la irradiación cutánea total con electrones combinada con poliquimioterapia frente al tratamiento conservador tópico, el tratamiento agresivo sólo consiguió unas tasas de remisiones completas más elevadas, pero los efectos secundarios fueron más importantes y no se observaron diferencias significativas con respecto a las supervivencias global o al tiempo libre de enfermedad24.

TABLA 4. ESTADIFICACIÓN TNMB DE LOS LINFOMAS CUTÁNEOS DE CÉLULAS T

TABLA 5. ESQUEMA TERAPÉUTICO DE LOS LINFOMAS CUTÁNEOS DE CÉLULAS T
Los tratamientos actuales implican una pauta terapéutica gradual, que se inicia con tratamientos conservadores. Las estrategias terapéuticas se dividen en dos categorías: tratamientos con un efecto cutáneo directo y tratamientos sistémicos. Los tratamientos con un efecto cutáneo directo son de primera elección en lesiones maculares, placas y en lesiones nodulares aisladas. Su eficacia clínica disminuye en las formas extensas (T2), y se obtienen unos porcentajes de remisiones completas (RC) inferiores al 50 %. En los pacientes con estadios avanzados suele ser necesario prescribir tratamientos sistémicos. Los tratamientos con un efecto cutáneo directo incluyen la fototerapia con PUVA (psoralenos junto a luz ultravioleta A), fototerapia con UVB (luz ultravioleta B), quimioterapia tópica con mecloretamina (mostaza nitrogenada) o carmustina (BCNU), gel de bexaroteno al 1 % (retinoide), radiación cutánea total (baño de electrones) y radioterapia convencional para lesiones cutáneas sintomáticas. Ningún estudio ha demostrado la superioridad de un tratamiento concreto de acción directa cutánea, y la decisión de escoger un tratamiento determinado depende de la experiencia del médico y de las preferencias del paciente7,19,25.
Tratamientos con un efecto cutáneo directo
Fototerapia. La fototerapia representa un tratamiento eficaz para los estadios precoces (T1/T2) de la micosis fungoide. El tratamiento con PUVA puede conseguir RC en el 65-95 % de los pacientes. También pueden conseguirse respuestas parciales (RP) en lesiones tumorales (T3) y en formas eritrodérmicas (T4) (estadios IIB y III). La fototerapia con UVB consigue RC en más del 80 % de los casos en lesiones T1 (IA) con una menor respuesta si las lesiones son más infiltradas. En estudios recientes, tanto la radiación UVB de banda estrecha (TL-01, 311 nm) como UVA1 (340-400 nm) son igualmente efectivas en micosis fungoides con lesiones incipientes. Cualquiera de estos tratamientos consigue una respuesta más limitada (60 %) en T2 (IB). Existe una cierta controversia con respecto a la necesidad de realizar tratamientos de mantenimiento en pacientes con micosis fungoide tras haber obtenido la resolución clínica de las lesiones. Una limitación habitual a la eficacia de la fototerapia son las áreas flexurales, que reciben una menor irradiación26-29.
Quimioterapia tópica. El tratamiento tópico quimioterápico representa una opción terapéutica eficaz en los estadios precoces de la enfermedad o como tratamiento adyuvante tras conseguir la remisión con fototerapia. La aplicación tópica de mecloretamina (mostaza nitrogenada a concentraciones de 0,01-0,02 % en pomada o ungüento) o BCNU consigue unos resultados similares o discretamente inferiores a los obtenidos con las radiaciones UV (80-90 % RP, 50-80 % RC en estadio IA). La frecuente aparición de dermatitis de contacto y reacciones de hipersensibilidad generalizadas puede limitar la utilización de la mecloretamina tópica. La BCNU es un fármaco con mejor tolerancia. Su aplicación continuada puede ocasionar leucopenia y atrofia cutánea con la aparición de telangiectasias en las zonas de aplicación. Ésta suele ser diaria durante un periodo variable de entre 2 y 12 meses, aunque tampoco hay pautas establecidas por falta de estudios controlados adecuados. Algunos estudios no han constatado diferencias significativas con respecto al placebo30,31.
Corticoides tópicos. Los corticoides tópicos potentes o en oclusión tienen resultados comparables en T1 (90 % RP; 60 % RC), pero su eficacia se reduce considerablemente cuando existen lesiones extensas (25 % RC en T2)32.
Retinoides tópicos. El bexaroteno al 1 % es un retinoide con eficacia más limitada en T1-T2 que los anteriores tratamientos, pero podría plantearse su empleo en formas refractarias y especialmente las formas poiquilodérmicas de micosis fungoides que aparecen en áreas flexurales. Su aplicación 2 veces al día consigue una respuesta cercana al 60 %. Produce cierto grado de irritación local que puede minimizarse mediante un incremento gradual de las aplicaciones. Su aplicación está contraindicada en el embarazo33.
Radioterapia. Los linfomas suelen ser tumores muy radiosensibles. La radioterapia con electrones aplicada en forma de radiación cutánea total (baño de electrones) es un tratamiento muy eficaz, especialmente en estadios iniciales (RC superiores al 90 % en estadios I y IIA). Es menos tóxica que la radioterapia convencional con fotones, y la radiación sólo alcanza la dermis papilar. Requiere personal sanitario e infraestructuras especializadas. En estudios comparativos no se ha conseguido demostrar una eficacia superior con respecto a los otros tratamientos de acción directa cutánea (PUVA, mecloretamina). Se utiliza fundamentalmente en pacientes con afectación cutánea difusa o extensa por placas gruesas e infiltradas (estadio IB)
o con múltiples tumores (IIB), y también en lesiones refractarias a otras opciones terapéuticas de acción cutánea local. En estos casos la tasa de remisiones completas alcanzada es mucho menor. Se ha considerado primera línea terapéutica en la micosis fungoide asociada a mucinosis folicular. Una pauta habitual consiste en alcanzar una exposición acumulada de 36 Gy (1,5-2 Gy/día) en un plazo aproximado de 10 semanas. Muchos pacientes toleran un segundo ciclo de radioterapia en caso de recaídas, aunque los mejores resultados y las respuestas más persistentes suelen obtenerse tras el primer ciclo de tratamiento. Una vez alcanzada la remisión, suele requerirse la prescripción de un tratamiento adyuvante (PUVA o quimioterapia tópica), ya que existe una gran probabilidad de recaídas. Es un tratamiento no exento de efectos secundarios cutáneos como alopecia, telangiectasias y aparición de epiteliomas cutáneos.
Debido a la toxicidad de la radioterapia externa con fotones de alto voltaje, se limita su uso a las áreas de micosis fungoide resistentes o al control de lesiones tumorales. Una pauta satisfactoria puede consistir en la administración de varias fracciones (2-3 fracciones de 400 cGy a 80-120 Kv)34,35.
Tratamientos sistémicos
Los tratamientos sistémicos incluyen diversas pautas de poliquimioterapia, fármacos citotóxicos y fármacos modificadores de la respuesta inmunitaria. De más reciente incorporación son un nuevo retinoide, el bexaroteno y el DAB (389)-IL-2 (interleucina-2-proteí-na diftérica de fusión) que ofrecen nuevas opciones terapéuticas para la fase avanzada.
Pautas de mono o poliquimioterapia. La quimioterapia sistémica se usa como tratamiento paliativo en las recaídas o en los casos refractarios a las terapias de acción directa sobre la piel o bien en pacientes con enfermedad avanzada. En general, los linfomas T son quimiorresistentes y sólo se consiguen respuestas parciales y de corta duración. Los tratamientos con un único fármaco sólo consiguen respuestas parciales (30-50 %), con una duración media inferior a 6 meses. No hay estudios que avalen un aumento de la supervivencia. Suelen utilizarse fármacos alquilantes como la mecloretamina, la ciclofosfamida o el clorambucilo,
o antimetabolitos como el metotrexato. Las tasas de respuesta son siempre menores a las obtenidas mediante combinaciones de quimioterápicos36.
Monoquimioterapia. El metotrexato se utiliza en algunos casos de micosis fungoide avanzada. Se administra a dosis semanales intramusculares o subcutáneas de 50 mg durante 2 a 4 meses. La ciclofosfamida, vinblastina o mecloretamina sistémica también están aprobadas por la Food and Drug Administration (FDA) en el tratamiento de la micosis fungoide avanzada. Estudios recientes investigan el papel de los análogos de las purinas (2-desoxicoformicina [pentostatina], 2-clordesoxiadenosina y fludarabina). También el clorambucilo, la gemcitabina, la bleomicina, el etopósido y la adriamicina se han utilizado como único tratamiento en casos aislados con una eficacia relativa37-40.
Poliquimioterapia. Se emplean en linfomas cutáneos T avanzados, refractarios o con afectación sistémica. En la micosis fungoide se han utilizado múltiples pautas de quimioterapia: CHOP (ciclofosfamida, vincristina, adriamicina, prednisona), CAVE (ciclofosfamida, adriamicina, vincristina, etopósido) o EPOCH (etopósido, vincristina, adriamicina, ciclofosfamida, prednisona). La mayoría de los pacientes desarrollarán una recaída al cabo de unos meses. No existen estudios aleatorizados que demuestren un aumento en la supervivencia con ningún tratamiento o pauta terapéutica36.
Fotoféresis. Este tratamiento está ideado para aquellos enfermos en los que se demuestra enfermedad en sangre periférica (detección de células de Sézary, reordenamiento clonal o citometría de flujo con ratio CD4+/CD7- aumentada). La fotoféresis o fotoquimioterapia extracorpórea, en estas circunstancias, ha conseguido respuestas superiores al 50 % en casos refractarios a otros tratamientos, aunque la mayor experiencia se tiene con el síndrome de Sézary. Esta técnica es más efectiva si se emplea en pacientes con un sistema inmunitario competente con células CD8+, CD56+ conservadas, es decir, que no hayan sido sometidos a quimioterapia previamente41.
Tratamientos inmunomoduladores.Interferones (IFN-α). El objetivo de la inmunoterapia es reestablecer el equilibrio de una respuesta inmunitaria antitumoral de patrón de secreción de citocinas Th1 y generación de linfocitos T citotóxicos. Algunos de los fármacos inmunomoduladores utilizados también han demostrado un efecto in vitro antiproliferativo. Consiguen una tasa de respuesta global de hasta el 80 % (sobre todo en estadios I y II) pero las remisiones completas son inferiores al 25 %. Posiblemente su utilización en pautas combinadas (con PUVA o fotoféresis) aumente la eficacia de la respuesta.
El IFN-α2a recombinante es un tratamiento eficaz para la micosis fungoide. Puede administrarse tanto por vía subcutánea, intramuscular o incluso intralesional. Suele iniciarse el tratamiento con dosis de entre 3-5 × 106 U/3-4 veces a la semana (o diario) durante 6-9 meses o más, con una respuesta clínica inicial objetivable durante los 2-5 meses primeros de tratamiento. Existe una relación dosis/respuesta, por lo que pueden aumentarse las dosis antes de abandonar el tratamiento por falta de eficacia. Considerando los casos con un elevado riesgo de recaída, se recomiendan dosis de mantenimiento, de incluso años, en aquellos pacientes con enfermedad inicial avanzada o con respuestas mantenidas. Los pacientes tratados con IFN-α recombinante durante largos periodos presentan un riesgo aumentado de desarrollar enfermedades autoinmunes como diabetes mellitus, tiroiditis o anemia hemolítica.
Entre los efectos secundarios relacionados con el empleo de IFN-α destacan los habituales de tipo gripal, depresión, alteración del recuento hematológico, aumento de las enzimas hepáticas, alteraciones tiroideas, cefalea, diarrea, etc. La administración nocturna del fármaco junto con la prescripción combinada de paracetamol puede hacer más llevadero el síndrome seudogripal acompañante. Algunos estudios recientes señalan una mayor eficacia del tratamiento combinado de IFN-α y PUVA, IFN-α y retinoides, IFN-α y desoxicoformicina, e IFN-α y bexaroteno. Sin embargo, de entre todas estas pautas combinadas, la asociación de IFN-α y PUVA parece ser la más eficaz42.
Existe un experiencia limitada en el tratamiento de la micosis fungoide con IFN-α natural (IFN-α2b) o con IFN-α. Los resultados obtenidos con estos fármacos parecen ser equiparables a los obtenidos con el IFN-α2a. Pueden utilizarse en casos refractarios al IFN recombinante. El estudio más extenso publicado de tratamiento con IFN-α en la micosis fungoide incluía 16 pacientes con enfermedad refractaria a otros tratamientos, obteniéndose una respuesta transitoria en 5 pacientes de una duración media de 10 meses. La dosis utilizada fue de 0,5 mg/m2 y los efectos secundarios fueron similares a los observados con el IFN-α2a 43,44
Retinoides. Existen varios estudios que evalúan la eficacia de los retinoides en diferentes estadios de micosis fungoides refractarias y otros linfomas de las células T no micosis fungoide. Los más empleados son el acitretín y, más recientemente, un nuevo retinoide aprobado por la FDA, el bexaroteno. Se emplean como único tratamiento o en combinación con otros tratamientos (PUVA o IFN-α). El mecanismo de acción fundamental residiría en su acción antiproliferativa promoviendo la apoptosis, regulando la proliferación celular, en un efecto inmunopotenciador, reduciendo la producción de citocinas Th2 y en una actividad antiinflamatoria. El bexaroteno posee una acción selectiva sobre el receptor X del retinoide (RXR). Suele administrarse en dosis de 300 mg/m2 diarios durante 3-5 meses. Suele ser un tratamiento eficaz en aproximadamente el 50 % de los casos. Suele utilizarse en casos refractarios, tanto en estadios precoces como avanzados. Las respuestas observadas parecen estar en relación a la dosis administrada.
Los efectos secundarios son asimismo dependientes de la dosis. El desarrollo de hipertrigliceridemia es un efecto secundario prácticamente constante que obliga a pautar fármacos hipolipemiantes (estatinas) previos al inicio del tratamiento. No debe emplearse gemfibrozilo, ya que puede aumentar los niveles de bexaroteno. Por todo ello, se aconseja realizar controles lipídicos periódicos (cada 15 días) al iniciar el tratamiento. Un hipotiroidismo central (reducción de hormona tiroestimulante y tiroxina libre) es otro efecto secundario frecuente. El hipotiroidismo debe monitorizarse con la determinación de tiroxina y puede tratarse con levotiroxina. Otros efectos secundarios ocasionales incluyen leucopenia, una alteración de las pruebas hepáticas y cefaleas persistentes. Todas estas alteraciones suelen ser reversibles tras la supresión del fármaco. Como en todos los retinoides, debe realizarse una anticoncepción efectiva debido al riesgo de malformaciones45.
Interleucina-2 (IL-2). La IL-2 es un factor de crecimiento del linfocito T, que aumenta la respuesta inmunitaria a través de la estimulación de células NK (natural killer), de células citotóxicas, la producción de IL-12 e IFN-α. La administración previa de factores que estimulan los linfocitos T como IFN-α o retinoides puede potenciar sus efectos. La experiencia de este fármaco en el tratamiento de la micosis fungoide es limitado y la tasa de respuestas poco relevante46.
Proteína de fusión IL-2-toxina diftérica (DAB389-IL-2, denileukin diftitox). Es una proteína recombinante de fusión de actividad citotóxica contra las células T que expresen el receptor de la IL-2 (CD25). La proteína une la región que conecta el receptor de la IL-2 con la toxina diftérica. Tras la unión con el receptor y la endocitosis celular, la toxina diftérica es liberada e inhibe la producción proteica celular. Este fármaco ha sido aprobado recientemente por la FDA en el tratamiento de las formas avanzadas refractarias de micosis fungoide. Sólo se utiliza en aquellos linfomas en los que al menos el 20 % de las células expresan el antígeno CD25.
El denileukin diftitox consigue una respuesta global del 30 %. Suele administrarse mediante perfusión intravenosa lenta de 9-18 g/kg/día durante 5 días consecutivos, repitiendo dicha pauta cada 3 semanas. La mayoría de los pacientes presentan una respuesta tras 2-6 ciclos. Los efectos secundarios descritos incluyen un síndrome seudogripal, náuseas, vómitos, hipotensión, edema e hipoalbuminemia, que suelen manifestarse aproximadamente 10 días después de iniciarse el tratamiento. Se han descrito asimismo reacciones de hipersensibilidad con hipotensión y dolor de espalda, y discretos aumentos de creatinina y transaminasas47,48.
Otros inmunomoduladores. Se ha descrito un amplio espectro de modalidades terapéuticas de carácter experimental, habitualmente a partir de series no aleatorizadas, que incluyen un número reducido de pacientes. Algunos estudios han valorado la eficacia de diversos anticuerpos monoclonales unidos o no a toxinas o elementos radiactivos en la micosis fungoide: anticuerpos anti-CD52 (CAMPATH-1H), anti-CD25 o anti-CD4. También existe una experiencia limitada en el tratamiento con nuevos inhibidores de la fosforilasa de los nucleósidos purínicos (peldesine), así como con la IL-2 subcutánea o intralesional o con los bloqueantes de receptores de linfocitos como los antiCD11, LFA-3TIP. Lo mismo ocurre con las vacunas an-titumorales49,50.
La interleucina-12 (IL-12) recombinante es un potente inductor del IFN-α de las células T y células NK. Estimula directamente los linfocitos citotóxicos e inhibe la producción de IL-4. Los linfomas T cutáneos avanzados presentan un defecto marcado de la producción de IL-12 probablemente como consecuencia de la producción de IL-10 por parte de las células malignas. En estudios en fase I, la IL-12 (subcutánea o intralesional) demostró cierta actividad con una respuesta global superior al 50 % en casos de micosis fungoide avanzada51.
Tratamientos combinados. La utilización de tratamientos combinados resulta una práctica habitual en la micosis fungoide. Se han empleado pautas combinadas de fotoféresis, fototerapia, IFN-α y retinoides en diferentes esquemas o protocolos. Sin embargo, el número de estudios aleatorizados es escaso y la significación de los resultados obtenidos difícil de valorar. En un estudio aleatorizado se demostró una mayor tasa de remisiones completas con la combinación de PUVA e IFN-α que con la asociación IFN-α y acitretín en una serie de pacientes en estadios I y II. En otro estudio se consiguió un mayor número de remisiones completas con PUVA y IFN-α en lesiones T1-T4, pero en el grupo T1-T2 no aportaba beneficio con respecto al tratamiento aislado sólo con PUVA. En contraste con el tratamiento con PUVA, la combinación de PUVA con retinoides parece conseguir remisiones más rápidas y con una menor dosis de UVA, aunque dichas remisiones no parecen ser más d uraderas. La asociación IFN-α con retinoides no parece aportar ningún efecto beneficioso. La adición de IFN-α tras un tratamiento con fotoquimioterapia extracorpórea podría potenciar los resultados del tratamiento, así como una triple terapia con IL-27,19,52.
Trasplante de médula ósea. Se ha utilizado de forma puntual el trasplante de médula ósea o de células madre en casos de micosis fungoide de evolución agresiva y refractarios a otros tratamientos. Un trabajo reciente ilustra la eficacia del trasplante de médula ósea alogénico enriquecido con células madre para conseguir una remisión clínica mayor y más duradera en casos en fases avanzadas si se compara con los casos en los que se ha realizado un trasplante autólogo. El efecto de un fenómeno de injerto contra huésped, en este caso injerto contra linfocito (neoplásico), parecería tener un papel importante, controlando el desarrollo de nuevas lesiones cutáneas53.
SÍNDROME DE SÉZARY
DiagnósticoAspectos clínicos
El síndrome de Sézary, la variante leucémica de la micosis fungoide, se manifiesta por la tríada de eritrodermia, linfadenopatías y más de 1.000 células linfoides atípicas circulantes (células de Sézary) por mm3. Dichas células son células mononucleares de tamaño moderadamente grande con núcleos convolutos (fig. 5). Los pacientes presentan un prurito generalizado de gran intensidad, refractario al tratamiento, ectropión, alopecia, distrofia ungueal, edema periférico, e hiperqueratosis palmoplantar. En la clasificación de la EORTC se considera como un linfoma cutáneo de células T de curso agresivo, progresa más rápidamente, y es más resistente al tratamiento que una micosis fungoide convencional. La médula ósea suele verse respetada a pesar del componente en sangre periférica54.
En un contexto clínico adecuado (prurito, eritrodermia, queratodermia, alopecia, distrofia ungueal, adenopatías y esplenomegalia) apoya el diagnóstico de síndrome de Sézary la presencia de más del 5-20 % de células de Sézary, población linfocitaria predominante CD4+/CD7- o ratio CD4/CD8 elevada por citometría de flujo o reordenamiento clonal en sangre periférica (100 % de casos por PCR). Este último elemento debe considerarse esencial en el diagnóstico de un síndrome de Sézary55.
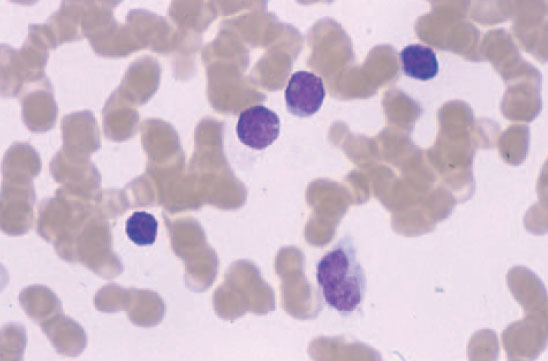
Fig. 5.—Células grandes de núcleos convolutos correspondientes a células de Sézary en sangre periférica.
Aspectos histopatológicos
Histológicamente, el síndrome de Sézary es indistinguible de la micosis fungoide de placa no infiltrada, a menudo con cambios sutiles e inespecíficos y poco epidermotropismo. El fenotipo predominante es CD3/ CD4+ (cooperador) aunque algunos casos expresan un fenotipo supresor/citotóxico CD3/CD8+ lo que suele conllevar una evolución más agresiva. En el síndrome de Sézary con frecuencia se detecta en las células linfoides atípicas un fenotipo aberrante CD3+, CD4+, CD7- (pérdida de CD7) o una relación CD4/CD8 elevada. Se detecta un clon linfoide T en la sangre periférica en casi el 100 % de los pacientes.
Aspectos pronósticos
En el síndrome de Sézary se han detectado como factores pronósticos adversos la presencia de inclusiones PAS+ citoplasmáticas en las células de Sézary, un fenotipo con pérdida del antígeno CD7 y la observación de células de Sézary grandes circulantes. En el síndrome de Sézary, la supervivencia global a los 5 años es aproximadamente del 10-20 %, si bien en aquellos pacientes que no presentan ningún factor pronóstico adverso puede aumentar al 60 % mientras que en aquellos con dos o tres factores pronósticos, la supervivencia se reduciría a menos del 5 %54,56.
Aspectos terapéuticos
La producción de citocinas con actividad inmunosupresora o patrón Th2 (IL-4, IL-10) por parte de las células neoplásicas contribuye a la progresión de la enfermedad, por lo que la conversión a un patrón de citocinas Th1 (IL-2, IFN) mediante reducción de la masa tumoral (quimioterapia, fotoféresis) o con tratamientos inmunomodulares como el IFN-α o los retinoides son de utilidad en el síndrome de Sézary. Las pautas agresivas de poliquimioterapia han resultado poco eficaces en este proceso. En los pacientes con un síndrome de Sézary, el tratamiento de elección o primera línea es un tratamiento conservador con fármacos citotóxicos en dosis bajas o mediante fotoféresis extracorpórea. El empleo de clorambucilo diariamente o en pulsos junto a prednisona en síndrome de Sézary (o micosis fungoide eritrodérmica) es de uso habitual57. También ha demostrado eficacia el tratamiento con metotrexato a dosis bajas58.
La fotoféresis extracorpórea consiste en una leucoféresis de leucocitos tratados con 8-metoxipsoraleno y expuestos a 1-2 J de UVA que luego se reinfunden al paciente. Estas células tratadas entran en apoptosis y además se produce una activación de monocitos circulantes a células dendríticas que procesan antígenos tumorales. La adición de factor estimulador de las colonias granulomonocíticas (GM-CSF) tras la fotoféresis en dosis de 125 g puede añadir algún beneficio. El GM-CSF busca aumentar la actividad presentadora de antígenos. La fotoféresis puede emplearse en casos refractarios con un régimen de aplicación de 2 días consecutivos al mes. Puede llegar a conseguir remisiones completas superiores al 50 %, sobre todo cuando el recuento CD8 es normal y el tiempo de evolución de la enfermedad es corto, siendo éstas más duraderas si se consigue la respuesta clínica en los primeros 6 meses. Los pacientes con un número elevado de células malignas o muy inmunodeprimidos habitualmente no responden a la fotoféresis como único tratamiento. En estos casos la asociación de IFN-α y bexaroteno parece más recomendable. Este tratamiento inmunoactivador combinado podría no sólo conseguir mejorías clínicas, sino también en datos de supervivencia. También puede asociarse crioterapia tópica, corticoides tópicos o radioterapia, así como PUVA que en algún trabajo reciente incluso apunta a un aumento de la supervivencia59. Recientemente la administración del anticuerpo monoclonal anti-CD52 o alemtuzumab (CAMPATH1H) parece obtener unos resultados alentadores en el control de la sintomatología de pacientes con síndrome de Sézary o formas de micosis fungoide avanzada particularmente eritrodérmicas60.
LINFOMAS CUTÁNEOS DE CÉLULAS T NO MICOSIS FUNGOIDE
Existe una variabilidad significativa en las manifestaciones clínicas de este grupo de linfomas cutáneos de células T. Las lesiones varían desde nódulos eritematosos o violáceos solitarios, nódulos agrupados o placas infiltradas, lesiones subcutáneas similares a paniculitis y múltiples pápulas infiltradas recurrentes y autoinvolutivas. El diagnóstico se establece mediante la combinación de las manifestaciones clínicas, histopatológicas, inmunohistoquímicas y de genética molecular. Como en todos los grupos de linfomas cutáneos, resulta fundamental una correcta correlación clinicopatológica. El estudio histopatológico evidencia un infiltrado dérmico difuso formado por células linfoides atípicas de tamaño variable que se extiende desde la dermis superficial al tejido celular subcutáneo. Suelen observarse numerosas mitosis. La epidermis suprayacente puede ulcerarse, aunque con frecuencia evidencia una intensa hiperplasia pseudoepiteliomatosa. Puede asimismo observarse epidermotropismo de intensidad variable. El estudio inmunofenotípico de las biopsias cutáneas permite definir la estirpe celular linfoide T (CD3+, CD2+, CD5+) y a menudo se evidencia la pérdida de alguno de los marcadores de células linfoides T maduras, y/o la expresión de marcadores característicos de activación linfoide, como el antígeno CD30. El análisis genotípico permite demostrar una población linfoide monoclonal de células T en la mayor parte de los casos.
La mayoría de los linfomas cutáneos de células T primarios distintos de la micosis fungoide presentan un similar algoritmo terapéutico. Los tumores solitarios o localizados pueden tratarse mediante su extirpación quirúrgica y/o radioterapia. Por otro lado, los pacientes con lesiones cutáneas multifocales o diseminadas suelen ser tratados habitualmente con poli-quimioterapia61,62.
PROLIFERACIONES LINFOIDES CUTÁNEAS CD30+
Las proliferaciones linfoides cutáneas CD30+ son un grupo de procesos linfoproliferativos cutáneos asociados a un pronóstico relativamente favorable, que poseen en común la positividad por parte de las células neoplásicas del antígeno Ki-1 o CD30, considerado inicialmente característico de las células de Reed-Sternberg de la enfermedad de Hodgkin. Este grupo incluye la papulosis linfomatoide, los linfomas de célula grande CD30+ (LC-CD30+) y ciertos procesos a los que se ha acordado denominar borderline, que corresponden a casos de discrepancia clinicohistológica para considerarlos como papulosis linfomatoide o como linfoma. Se considera que representan un espectro continuo de lesiones cuyos extremos serían la papulosis linfomatoide y el linfoma CD30+, respectivamente. La supervivencia media de este grupo es superior a 90 % a los 5 años (100 % en el caso de la papulosis linfomatoide, algo menor si se considera aisladamente a los LC-CD30+)63,64.
Papulosis linfomatoideDiagnóstico
La papulosis linfomatoide es un proceso linfoproliferativo indolente cuya histología simula una neoplasia linfoide maligna. Aunque es más frecuente en adultos, puede observarse a cualquier edad. Las lesiones consisten en pápulas o nódulos dérmicos que presentan una tendencia a la necrosis central y ulceración. Las lesiones involucionan de manera espontánea dejando cicatrices de aspecto varioliforme. Por lo general, se localizan en el tronco y en las extremidades, pueden aparecer agrupadas o tener una distribución múltiple, con número y tamaño variables, y es característico que estén en diferentes estadios evolutivos. El curso es crónico y recurrente. La involución de las lesiones suele observarse en 1 o 2 meses, con la aparición de nuevos elementos durante meses o años (fig. 6A).
En la mayor parte de los casos, la histología de la papulosis linfomatoide muestra un infiltrado linfoide de disposición perivascular e interfase con una disposición o morfología «en cuña». La presencia de células linfoides atípicas, de gran tamaño, parecidas a las células de Reed-Sternberg es característica. Dichas células son intensamente positivas para el antígeno CD30, expresado con un patrón perinuclear. También suele observarse un abundante infiltrado inflamatorio acompañante compuesto principalmente por polimorfonucleares neutrófilos y eosinófilos. No es infrecuente encontrar una hiperplasia epidérmica pseudocarcinomatosa suprayacente (fig. 6B). Este aspecto histológico clásico puede plantear el diagnóstico diferencial con la pitiriasis liquenoide o picaduras de artrópodo. Un pequeño porcentaje de pacientes con papulosis linfomatoide (entre el 5 al 20 % según los estudios), puede desarrollar a lo largo de la evolución un verdadero linfoma cutáneo (micosis fungoide o LC-CD30+) o asociarse a linfoma de Hodgkin. Parece probable que exista un origen clonal común para las células neoplásicas de pacientes afectados de papulosis linfomatoide, LC-CD30, micosis fungoide y linfoma de Hodgkin (dada la estrecha relación existente entre estas entidades). Las células neoplásicas tienen un fenotipo T cooperador (CD3+, CD4+, CD8- ) con presencia de CD30 en las grandes células atípicas (fig. 6C). Se puede detectar clonalidad mediante reordenamiento del RCT en el 50-70 % de los casos de papulosis linfomatoide. No se han descrito alteraciones genéticas específicas65-67.
Tratamiento
El tratamiento de la papulosis linfomatoide vendrá condicionado por la extensión del proceso cutáneo y por las molestias que pueda experimentar el paciente. En muchas ocasiones no se prescribe tratamiento y la simple explicación de la naturaleza del proceso por parte del médico es suficiente. En casos de lesiones necróticas o con tendencia a dejar cicatrices puede prescribirse tratamiento con fototerapia (UVB o PUVA) o metotrexato oral en dosis bajas (7,5 a 15 mg/semana)64,65. Estos tratamientos pueden bloquear la aparición de nuevos brotes. Sin embargo, las lesiones suelen recurrir a suspenderlos.
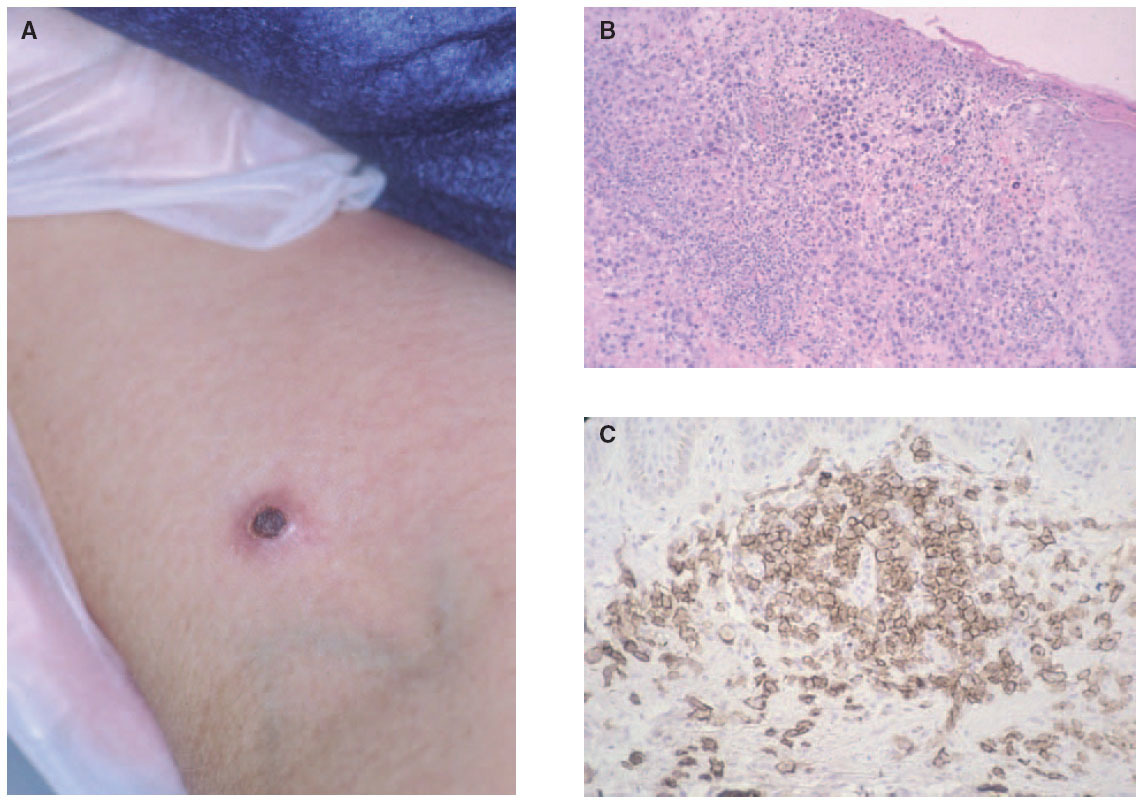
Fig. 6.—A) Pápula necrótica correspondiente a una lesión de papulosis linfomatoide. B) Infiltrado dérmico en cuña con necrosis epidérmica y células aisladas grandes de morfología atípica. (Hematoxilina-eosina, ×100.) C) Expresión profusa del antígeno CD30 en las células atípicas.
Linfoma primario cutáneo CD30+Dentro del espectro de procesos cutáneos primarios CD30+, en el polo opuesto a la papulosis linfomatoide, se encuentran los linfomas de célula grande CD30+. Desde el punto de vista clínico, su presentación más característica es en la forma de una única lesión tumoral, a veces con otras adyacentes de menor tamaño (fig. 7A). Ocasionalmente, las lesiones pueden presentar una regresión espontánea completa o parcial, aunque son frecuentes las recurrencias cutáneas. La diseminación extracutánea se detecta en alrededor del 10 % de los pacientes, principalmente a los ganglios linfáticos regionales y habitualmente en pacientes con una enfermedad cutánea multicéntrica64,68.
Histopatología
Histológicamente se caracteriza por una proliferación densa de células linfoides grandes de citoplasma poligonal y morfología atípica al menos el 30 % del infiltrado son células neoplásicas (fig. 7B). El fenotipo suele ser T CD4/CD45RO. La positividad para CD30 en estos casos sueles ser muy marcada, con más del 75 % de las células neoplásicas (fig. 7C). Existe una variante no anaplásica de este tumor, que supone un 20 % de los casos, en la que el infiltrado, de aspecto pleomórfico, está compuesto por células de tamaño mediano y grande de núcleo irregular o de aspecto inmunoblástico. No parecen existir diferencias en el comportamiento clínico de los linfomas CD30+ respecto a si son morfológicamente anaplásicos, pleomórficos o inmunoblásticos (no anaplásicos). Se ha descrito la coexpresión de CD56 (antígeno de células NK) en algunos casos, quizá siguiendo un comportamiento más agresivo69. Sin embargo, no es infrecuente la expresión de marcadores citotóxicos (TIA-1, granzyme B), sin que ello parezca afectar al pronóstico.
En los linfomas primarios cutáneos CD30+ no se detecta la translocación cromosómica t(2;5)(p23;q35), que caracteriza a los linfomas de origen nodal, ni por tanto la expresión de su proteína de fusión NPM/ALK (anaplastic lymphoma kinase)70. Tampoco expresan EMA y son CD15- a diferencia de los linfomas ganglionares y del linfoma de Hodgkin. Es habitual detectar re-ordenamiento del RCT.
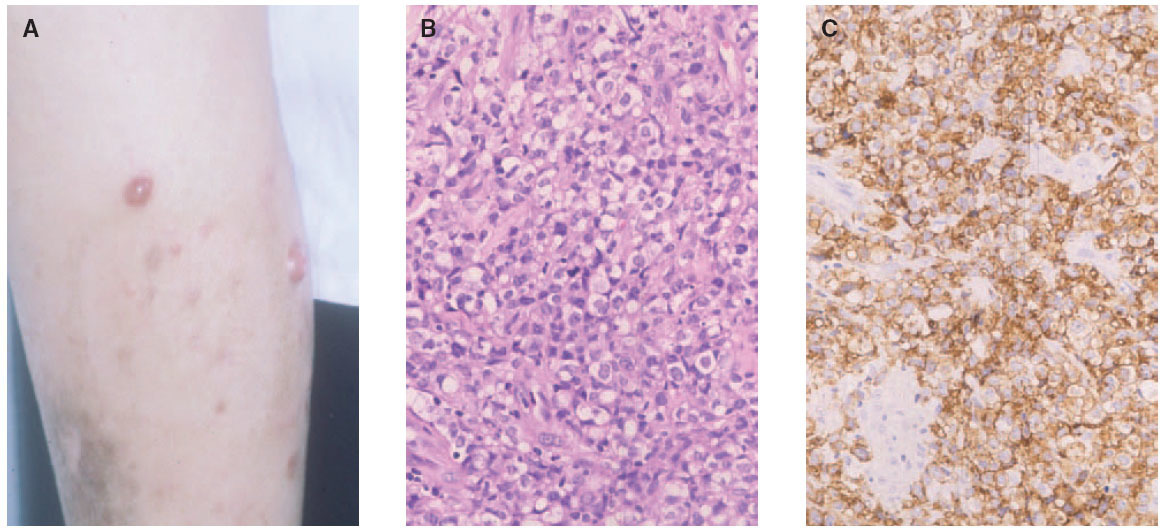
Fig. 7.—A) Lesiones nodulotumorales agrupadas correspondientes a un linfoma cutáneo de células grandes CD30+. B) Infiltrado denso, cohesivo, de células grandes atípicas. (Hematoxilina-eosina, ×200.) C) Marcada expresión del antígeno CD30 en las células neoplásicas.
Manejo terapéutico
El pronóstico de estos tumores es muy favorable, aunque exista extensión ganglionar secundaria locorregional (supervivencia a los 5 años superiores al 75-95 % según las series). En un reciente estudio multicéntrico europeo se detectó extensión extracutánea en un porcentaje relativamente bajo de pacientes, a pesar de lo cual, ante dicha posibilidad se aconseja un estudio de extensión que incluya un examen de médula ósea, una citología de sangre periférica y estudios de imagen adecuados64. El tratamiento más indicado es difícil de establecer debido al escaso número de estudios controlados de que se dispone; sin embargo, debe ser conservador mediante cirugía o radioterapia
o bien mo noquimioterapia con metotrexato en lesiones localizadas. La afectación cutánea extensa multicéntrica debe plantear la posibilidad de empleo de poliquimioterapia. En algunos casos se ha observado una involución espontánea del tumor (tabla 4).
LINFOMAS PRIMARIOS CUTÁNEOS CD30-
Linfoma primario cutáneo de células T grandes CD30-Los linfomas primarios cutáneos de célula grande T que no expresan el antígeno CD30 y que se desarrollan en pacientes que no tienen antecedentes de micosis fungoide, representan un grupo particular reconocido en la EORTC que siguen habitualmente un curso evolutivo rápido y agresivo. Se presentan inicialmente como lesiones nodulares tumorales únicas o localizadas, o bien en forma de placas infiltradas y nódulos múltiples.
La histología muestra un infiltrado nodular o difuso con epidermotropismo mínimo o inexistente. Las células son de mediano o gran tamaño y presentan unos núcleos pleomórficos. Las células grandes neoplásicas representan, al menos, el 30 % del total de la población tumoral. El aspecto histológico de estos tumores puede ser idéntico a micosis fungoide transformadas en linfomas difusos de células grandes (fig. 8). El inmunofenotipo es CD3+/CD4+/CD8- con pérdida de antígenos pan-T (CD5, CD7). No hay descritas anomalías genéticas específicas. Se detecta habitualmente un reordenamiento monoclonal del RCT.
El tratamiento de elección, considerando su evolución agresiva, es la poliquimioterapia; sin embargo, la supervivencia a los 5 años es baja, de alrededor del 12 %. En los casos localizados puede iniciarse un tratamiento más conservador mediante radioterapia71.
Linfoma primario cutáneo de células T pleomórficas de pequeño/mediano tamañoLos linfomas primarios cutáneos de células T pleomórficas de pequeño/mediano tamaño suponen un diagnóstico de exclusión cuando se ha descartado micosis fungoide en fase tumoral (especialmente si está asociada a mucinosis folicular), síndrome de Sézary y linfoma subcutáneo tipo paniculítico, así como con los pseudolinfomas T. La observación de un fenotipo aberrante y la demostración de clonalidad linfoide T apoyarían el diagnóstico de linfoma mientras que la presencia de células CD8, CD20 e histiocitos sugerirían un pseudolinfoma62,72.
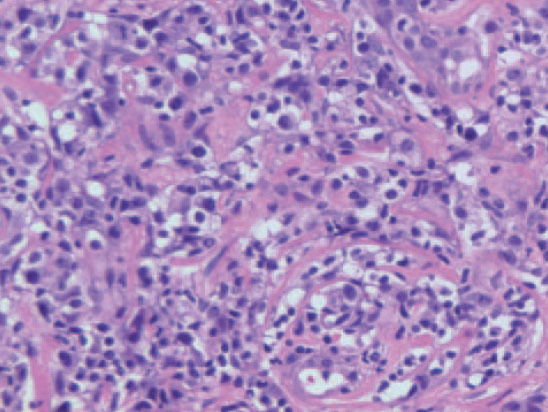
Fig. 8.—Infiltrado dérmico por células grandes que muestran pleomorfismo nuclear en un caso de linfoma cutáneo pleomórfico de células grandes CD30 negativo. (Hematoxilina-eosina, ×200.)
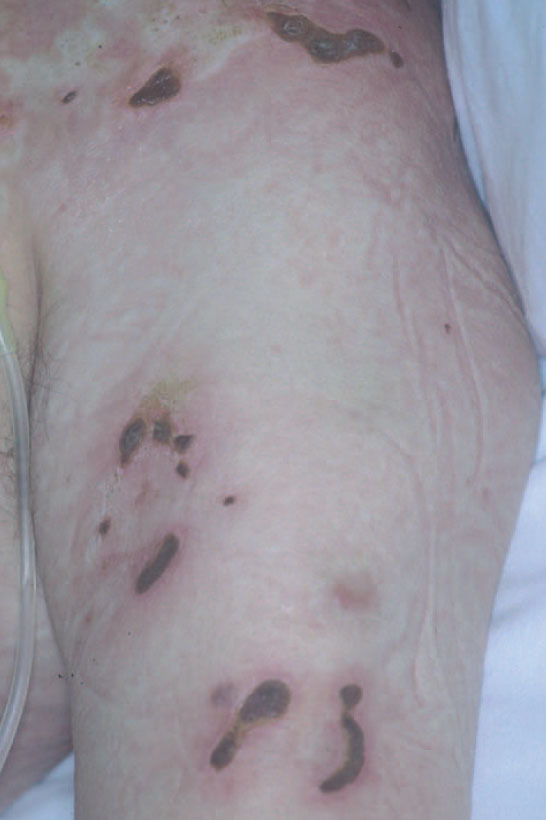
Fig. 9.—Placas diseminadas con tendencia a la ulceración en un paciente con linfoma cutáneo tipo paniculítico.
Estos linfomas se presentan como uno o varios nódulos eritematosos o rojo-purpúricos. La histología muestra un infiltrado denso difuso o nodular de células pleomórficas pequeñas/medianas, localizado en la dermis superficial y profunda, con tendencia a invadir el tejido subcutáneo y escaso o nulo epidermotropismo. El infiltrado es bastante monomorfo con escasas células acompañantes. La diferenciación con el grupo de linfomas CD30- de célula grande se basa en la presencia de menos del 30 % de células grandes. Según algunos trabajos, parece que el pronóstico es mejor (supervivencia del 45 % a los 5 años) respecto de los linfomas de célula grande, por lo que son clasificados provisionalmente como grupo diferente a éstos en la clasificación de la EORTC.
El fenotipo habitual es CD3/CD4+/CD5+/CD7- , y con frecuencia CD8- . Los linfomas con fenotipo CD4+/CD56+ sin otros marcadores T parecen tener peor pronóstico. El reordenamiento del RCT es habitualmente monoclonal. En formas localizadas se emplea inicialmente radioterapia. En caso de lesiones numerosas se requieren pautas de mono/poliquimio-terapia que incluya ciclofosfamida o IFN-α.
LINFOMA T SUBCUTÁNEO TIPO PANICULITIS
Es un linfoma cutáneo primario poco frecuente que clínicamente se manifiesta como nódulos subcutáneos y placas infiltradas que sugieren el diagnóstico de paniculitis. Las lesiones suelen localizarse en las piernas y pueden asociarse a sintomatología sistémica secundaria a un síndrome hemofagocítico con pancitopenia, fiebre y hepatosplenomegalia. La diseminación ganglionar y visceral es poco frecuente. Suelen seguir una evolución agresiva con una supervivencia corta, debido al intenso síndrome hemofagocítico. Sin embargo, parece existir un subgrupo de pacientes que presentan un curso indolente con afectación exclusivamente cutánea y recurrente durante largos periodos de tiempo (fig. 9).
Histológicamente se observa un infiltrado linfoide atípico con un patrón difuso limitado al tejido celular subcutáneo de características similares a una paniculitis lobulillar. Las células linfoides son de características pleomórficas de tamaño pequeño o intermedio. Pueden observarse, asimismo, linfocitos grandes con núcleos irregulares e hipercromáticos y abundantes mitosis. De forma ocasional puede observarse una fagocitosis de los detritos nucleares y de fragmentos de hematíes por parte de histiocitos reactivos (hemofagocitosis). Esta observación parece ser un signo con valor pronóstico negativo. Con frecuencia se observa un patrón de destrucción de los adipocitos marginales característico, con invasión del panículo por las células neoplásicas. Son frecuentes la infiltración y lesiones vasculares, que provocan amplias zonas de necrosis (angiocentricidad) (fig. 10).
El fenotipo habitual es CD8+ (fenotipo citotóxico). Las células muestran positividad para gránulos citotóxicos (TIA-1, granzyme B, perforina). No se asocia con una infección por el virus de Epstein-Barr (VEB). De forma ocasional puede demostrarse la expresión de marcadores de células NK (CD56, CD57, CD16), lo que parece conllevar un peor pronóstico, asociándose a hemofagocitosis. Sin embargo, de forma constante, las células expresan antígenos pan T (CD2, CD3), lo cual permite diferenciarlos de los verdaderos linfomas de células NK. Suele demostrarse la existencia de un reordenamiento monoclonal de la cadena y/o del receptor de la célula T73-75. El diagnóstico diferencial clínico incluye las lesiones nodulares inflamatorias de las extremidades inferiores como las paniculitis lobulillares, las vasculitis de vaso de tamaño intermedio y los procesos infecciosos. Muchos de los casos descritos de paniculitis histiocítica citofágica y de enfermedad de Weber-Christian probablemente correspondan a esta entidad. El linfoma subcutáneo tipo paniculitis es característicamente refractario tanto a la quimioterapia (mitoxantrone, fludarabina y dexametasona) como a la radioterapia. El pronóstico suele ser malo, con una supervivencia media inferior a los 3 años73,76.
Se ha descrito recientemente un grupo de linfoma T de célula grande epidermotropo con un inmunofenotipo característico citotóxico CD3- /CD7- /CD8+ y CD2- /CD4- /CD5- /CD30- con expresión de gránulos citotóxicos y mal pronóstico similar a los linfomas pleomórficos de célula grande CD30- 77 (tabla 6).
LINFOMA DE CÉLULAS NK
Los linfomas de células NK o linfoma tipo nasal NK (porque originalmente fueron descritos en la cavidad nasal) son un grupo de linfomas de observación excepcional. Son linfomas definidos por la expresión del antígeno CD56 (antígeno NK) y que, en ocasiones, también pueden expresar marcadores linfoides T (T/NK)78. Presentan una vinculación estrecha con el VEB, especialmente aquellos que se localizan en la cavidad nasal y con menos relevancia cuando se desarrollan primariamente en otras localizaciones como la piel. Este grupo de linfomas parece ser más prevalente en países asiáticos y centroamericanos, donde se ha descrito un cuadro clínico que simula a una hydroa vacciniforme. La evolución clínica de los linfomas angiocéntricos suele ser agresiva con frecuentes recurrencias, una rápida progresión y afectación extracutánea (ganglios linfáticos, médula ósea, bazo y nasofaringe). Sólo excepcionalmente se han descrito casos de evolución indolente79.
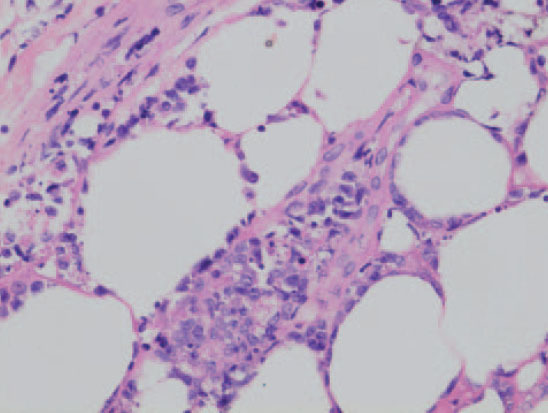
Fig.10.—Linfoma cutáneo tipo paniculitis, con un infiltrado del panículo por células linfoides pleomórficas con fenómenos de hemofagocitosis. (Hematoxilina-eosina, ×400.)
Los linfomas de células NK se han denominado asimismo linfomas angiocéntricos por la disposición arquitectural del infiltrado dérmico alrededor de los vasos y su carácter angiodestructivo. Sin embargo, la marcada destrucción vascular y la necrosis secundaria no es un elemento imprescindible para el diagnóstico. Por otro lado, el patrón morfológico de angiocentricidad no es exclusivo de este subgrupo de linfomas y puede también verse en otros linfomas, como el linfoma tipo paniculitis. Pueden presentar una imagen histopatológica variable observándose en la mayor parte de los casos una infiltración directa y destrucción vascular por linfocitos, los cuales pueden ser escasos en número, de pequeño tamaño y con un infiltrado celular mixto acompañante que puede plantear dudas diagnósticas con diversos procesos inflamatorios, sobre todo si hay poca atipia celular. El aspecto citológico es pleomórfico, con núcleos irregulares no cerebriformes. Ultraestructuralmente las células NK presentan gránulos citoplasmáticos que pueden ser ocasionalmente visibles también mediante tinciones de Giemsa o Wright. El inmunofenotipo más característico es CD2- , CD56+, CD3+ citoplasmático con expresión variable de otros antígenos de células NK como los antígenos CD16 y CD57. Las proteínas de gránulos citotóxicos pueden ser positivas, mientras que otros marcadores T son habitualmente negativos. Sin embargo, algunos linfomas tienen expresión variable de antígenos T (CD2, CD3, CD4, CD5, CD7 y CD8) por lo que se denominan linfomas NK-like o T-NK-like. Dicho grupo probablemente represente un subtipo particular, ya que de forma no excepcional desarrollan una infiltración de la médula ósea. En ambos fenotipos se ha demostrado VEB, aunque parece mucho más frecuente en los linfomas angiocéntricos-NK con respecto a los linfomas angiocéntricos predominio T. Se ha descrito asimismo la coexpresión de CD30 en algunos linfomas NK. Los linfomas de células NK no tienen un reordenamiento clonal del receptor de la célula T, aunque puede detectarse en aquellos linfomas con fenotipo T-NK.

TABLA 6. ESPECTRO DE LINFOMAS T CUTÁNEOS NO MICOSIS FUNGOIDE CON FENOTIPO CITOTÓXICO Y NK
El diagnóstico diferencial principal debe realizarse con los linfomas subcutáneos tipo paniculítico, ya que éstos también pueden presentar un patrón angiocéntrico y expresar ocasionalmente el antígeno CD5680. Los linfomas angiocéntricos NK se han tratado con diferentes pautas de poliquimioterapia. Sin embargo, no se ha demostrado que ninguno de estos tratamientos modifique el curso de la enfermedad. Las lesiones localizadas pueden tratarse inicialmente con radioterapia. La supervivencia es inferior al 50 % a los 5 años81.
El linfoma blástico de células NK es una neoplasia cutánea de estirpe no completamente aclarada. Su diagnóstico sólo puede establecerse ante la ausencia de expresión del antígeno CD33 y de mieloperoxidasa, descartando una posible infiltración cutánea por un proceso mieloproliferativo. La expresión de lisozima y del antígeno CD68 sugiere el diagnóstico de lesiones cutáneas secundarias a una leucemia mielomonocítica.
CONCLUSIONES
La micosis fungoide es el tipo más frecuente de linfoma cutáneo primario. La mayoría de los pacientes suele presentar lesiones correspondientes a las formas iniciales del proceso (T1, T2) y sólo un porcentaje reducido de pacientes fallecen de la enfermedad. El desarrollo de lesiones nodulares y tumorales en la micosis fungoide y los linfomas T no micosis fungoide, sin embargo, suponen un mal pronóstico (excepto en el caso de los procesos linfoproliferativos CD30+). La evaluación adecuada de un paciente con un linfoma cutáneo requiere la valoración detallada de la información clínica y los aspectos histológicos, inmunofenotípicos y moleculares. Resulta obligada la exclusión de un proceso linfoide sistémico mediante la realización de las oportunas exploraciones complementarias oportunas, incluyendo examen de médula ósea en algunas circunstancias, como en el caso de los linfomas CD30+ o en los linfomas de células NK. Existe un amplio espectro de opciones terapéuticas, pero no hay estudios aleatorizados y controlados que demuestren su eficacia y efecto sobre la supervivencia de los pacientes. Este hecho motiva que el tratamiento de este grupo de entidades, en muchas circunstancias, dependa de la experiencia de los distintos centros de referencia. La aproximación terapéutica en los linfomas cutáneos no micosis fungoide suele ser muy similar. Cuando existen lesiones solitarias o localizadas se recomienda un tratamiento mediante radioterapia o cirugía. Sin embargo, las formas cutáneas extensas o con diseminación extracutánea deben tratarse con mono/poliquimioterapia asociada en ocasiones a radioterapia, según la agresividad del tumor.
Fernando Gallardo. Servicio de Dermatología. Hospital del Mar.
P.º Marítimo, 25-29. 08003 Barcelona. España.
Fgallardo@imas.imim.es
Recibido el 18 de marzo de 2004.
Aceptado el 19 de mayo de 2004.



